PROBLEMS
PROBLEMS
Question 3.1
Valuable reagents. The following reagents are often used in protein chemistry:
|
CNBr |
Trypsin |
|
Urea |
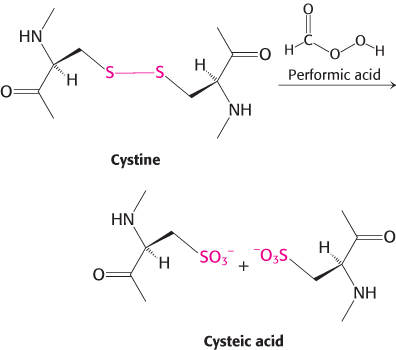
Performic acid |
|
Mercaptoethanol |
6 N HCl |
|
Chymotrypsin |
Phenyl isothiocyanate |
Which one is the best suited for accomplishing each of the following tasks?
Determination of the amino acid sequence of a small peptide.
Reversible denaturation of a protein devoid of disulfide bonds. Which additional reagent would you need if disulfide bonds were present?
Hydrolysis of peptide bonds on the carboxyl side of aromatic residues.
Cleavage of peptide bonds on the carboxyl side of methionines.
Hydrolysis of peptide bonds on the carboxyl side of lysine and arginine residues.
Question 3.2
The only constant is change. Explain how two different cell types from the same organism will have identical genomes but may have vastly divergent proteomes.
Question 3.3
Crafting a new breakpoint. Ethyleneimine reacts with cysteine side chains in proteins to form S-aminoethyl derivatives. The peptide bonds on the carboxyl side of these modified cysteine residues are susceptible to hydrolysis by trypsin. Why?
Question 3.4
Spectrometry. The absorbance A of a solution is defined as
A = log10(I0/I)
in which I0 is the incident-
A = εlc
The absorption coefficient of myoglobin at 580 nm is 15,000 M −1 cm−1. What is the absorbance of a 1 mg ml−1 solution across a 1-
Question 3.5
It’s in the bag. Suppose that you precipitate a protein with 1 M(NH4)2SO4 and that you wish to reduce the concentration of the (NH4)2SO4. You take 1 ml of your sample and dialyze it in 1000 ml of buffer. At the end of dialysis, what is the concentration of (NH4)2SO4 in your sample? How could you further lower the (NH4)2SO4 concentration?
Question 3.6
Too much or not enough. Why do proteins precipitate at high salt concentrations? Although many proteins precipitate at high salt concentrations, some proteins require salt to dissolve in water. Explain why some proteins require salt to dissolve.
Question 3.7
A slow mover. Tropomyosin, a 70-
Question 3.8
Sedimenting spheres. What is the dependence of the sedimentation coefficient s of a spherical protein on its mass? How much more rapidly does an 80-
Question 3.9
Frequently used in shampoos. The detergent sodium dodecyl sulfate (SDS) denatures proteins. Suggest how SDS destroys protein structure.
Question 3.10
Size estimate. The relative electrophoretic mobilities of a 30-
103
Question 3.11
Unexpected migration. Some proteins migrate anomalously in SDS-
Question 3.12
Sorting cells. Fluorescence-
Question 3.13
Column choice. (a) The octapeptide AVGWRVKS was digested with the enzyme trypsin. Which method would be most appropriate for separating the products: ion-
Question 3.14
Power(ful) tools. Monoclonal antibodies can be conjugated to an insoluble support by chemical methods. Explain how these antibody-
Question 3.15
Assay development. You wish to isolate an enzyme from its native source and need a method for measuring its activity throughout the purification. However, neither the substrate nor the product of the enzyme-
Question 3.16
Making more enzyme? In the course of purifying an enzyme, a researcher performs a purification step that results in an increase in the total activity to a value greater than that present in the original crude extract. Explain how the amount of total activity might increase.
Question 3.17
Divide and conquer. The determination of the mass of a protein by mass spectrometry often does not allow its unique identification among possible proteins within a complete proteome, but determination of the masses of all fragments produced by digestion with trypsin almost always allows unique identification. Explain.
Question 3.18
Know your limits. Which two amino acids are indistinguishable in peptide sequencing by the tandem mass spectrometry method described in this chapter and why?
Question 3.19
Protein purification problem. Complete the following table.
|
Purification Procedure |
Total protein (mg) |
Total Activity (units) |
Specific activity (units mg−1) |
Purification level |
Yield (%) |
|
|---|---|---|---|---|---|---|
|
Crude extract |
20,000 |
4,000,000 |
|
1 |
100 |
|
|
(NH4)2SO4 precipitation |
5,000 |
3,000,000 |
|
|
|
|
|
DEAE- |
1,500 |
1,000,000 |
|
|
|
|
|
Gel- |
500 |
750,000 |
|
|
|
|
|
Affinity chromatography |
45 |
675,000 |
|
|
|
|
Question 3.20
Part of the mix. Your frustrated colleague hands you a mixture of four proteins with the following properties:
|
|
Isoelectric point (pI) |
Molecular weight (in kDa) |
|---|---|---|
|
Protein A |
4.1 |
80 |
|
Protein B |
9.0 |
81 |
|
Protein C |
8.8 |
37 |
|
Protein D |
3.9 |
172 |
(a) Propose a method for the isolation of Protein B from the other proteins. (b) If Protein B also carried a His tag at its N-
Question 3.21
The challenge of flexibility. Structures of proteins comprising domains separated by flexible linker regions can be quite difficult to solve by x-
Chapter Integration Problems
Question 3.22
Quaternary structure. A protein was purified to homogeneity. Determination of the mass by gel-
Question 3.23
Helix–
Question 3.24
Peptide mass determination. You have isolated a protein from the bacterium E. coli and seek to confirm its identity by trypsin digestion and mass spectrometry. Determination of the masses of several peptide fragments has enabled you to deduce the identity of the protein. However, there is a discrepancy with one of the peptide fragments, which you believe should have the sequence MLNSFK and an (M+H)+ value of 739.38. In your experiments, you repeatedly obtain an (M+H)+ value of 767.38. What is the cause of this discrepancy and what does it tell you about the region of the protein from which this peptide is derived?
104
Question 3.25
Peptides on a chip. Large numbers of different peptides can be synthesized in a small area on a solid support. This high-
Question 3.26
Exchange rate. The amide hydrogen atoms of peptide bonds within proteins can exchange with protons in the solvent. In general, amide hydrogen atoms in buried regions of proteins and protein complexes exchange more slowly than those on the solvent-
Data Interpretation Problems
Question 3.27
Protein sequencing 1. Determine the sequence of hexapeptide on the basis of the following data. Note: When the sequence is not known, a comma separates the amino acids (Table 3.3).
Amino acid composition: (2R,A,S,V,Y)
N-
Trypsin digestion: (R,A,V) and (R,S,Y)
Carboxypeptidase digestion: No digestion.
Chymotrypsin digestion: (A,R,V,Y) and (R,S)
Question 3.28
Protein sequencing 2. Determine the sequence of a peptide consisting of 14 amino acids on the basis of the following data.
Amino acid composition: (4S,2L,F,G,I,K,M,T,W,Y)
N-
Carboxypeptidase digestion: L
Trypsin digestion: (3S,2L,F,I,M,T,W) (G,K,S,Y)
Chymotrypsin digestion: (F,I,S) (G,K,L) (L,S) (M,T) (S,W) (S,Y)
N-
Cyanogen bromide treatment: (2S,F,G,I,K,L,M*,T,Y) (2S,L,W)
M*, methionine detected as homoserine
Question 3.29
Applications of two-
Consider the following experiment: You suspect that a protein containing three cysteine residues has a single disulfide bond. You digest the protein with trypsin and subject the mixture to electrophoresis along one end of a sheet of paper. After treating the paper with performic acid, you subject the sheet to electrophoresis in the perpendicular direction and stain it with a reagent that detects proteins. How would the paper appear if the protein did not contain any disulfide bonds? If the protein contained a single disulfide bond? Propose an experiment to identify which cysteine residues form the disulfide bond.