14.5 Defects in Signal-Transduction Pathways Can Lead to Cancer and Other Diseases
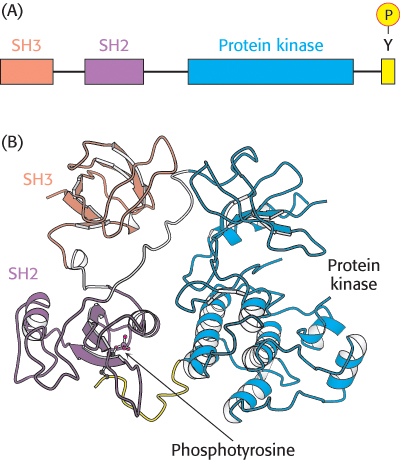
Figure 14.33: Src structure. (A) Cellular Src includes an SH3 domain, an SH2 domain, a protein kinase domain, and a carboxyl-terminal tail that includes a key tyrosine residue. (B) Structure of c-Src in an inactivated form with the key tyrosine residue phosphorylated. Notice how the three domains work together to keep the enzyme in an inactive conformation: the phosphotyrosine residue is bound in the SH2 domain and the linker between the SH2 domain and the protein kinase domain is bound by the SH3 domain.
[Drawn from 2PTK.pdb.]
 In light of their complexity, it comes as no surprise that signal -transduction pathways occasionally fail, leading to disease states. Cancer, a set of diseases characterized by uncontrolled or inappropriate cell growth, is strongly associated with defects in signal-transduction proteins. Indeed, the study of cancer, particularly cancers caused by certain viruses, has contributed greatly to our understanding of signal-transduction proteins and pathways.
In light of their complexity, it comes as no surprise that signal -transduction pathways occasionally fail, leading to disease states. Cancer, a set of diseases characterized by uncontrolled or inappropriate cell growth, is strongly associated with defects in signal-transduction proteins. Indeed, the study of cancer, particularly cancers caused by certain viruses, has contributed greatly to our understanding of signal-transduction proteins and pathways.
For example, Rous sarcoma virus is a retrovirus that causes sarcoma (a cancer of tissues of mesodermal origin such as muscle or connective tissue) in chickens. In addition to the genes necessary for viral replication, this virus carries a gene termed v-src. The v-src gene is an oncogene; it leads to the generation of cancerlike characteristics in susceptible cell types. The protein encoded by the v-src gene, v-Src, is a protein tyrosine kinase that includes SH2 and SH3 domains. The v-Src protein is similar in amino acid sequence to a protein normally found in chicken-muscle cells referred to as c-Src (for cellular Src; Figure 14.33A). The c-src gene does not induce cell transformation and is termed a proto-oncogene, referring to the fact that this gene, when mutated, can be converted into an oncogene. The protein that it encodes is a signal-transduction protein that regulates cell growth.
Why is the biological activity of the v-Src protein so different from that of c-Src? c-Src contains a key tyrosine residue near its C-terminal end that, when phosphorylated, is bound intramolecularly by the upstream SH2 domain (Figure 14.33B). This interaction maintains the kinase domain in an inactive conformation. However, in v-Src, the C-terminal 19 amino acids of c-Src are replaced by a completely different stretch of 11 amino acids that lacks this critical tyrosine residue. Thus, v-Src is always active and can promote unregulated cell growth. Since the discovery of Src, many other mutated protein kinases have been identified as oncogenes.
The gene encoding Ras, a component of the EGF-initiated pathway, is one of the genes most commonly mutated in human tumors. Mammalian cells contain three 21-kDa Ras proteins (H-, K-, and N-Ras), each of which cycles between inactive GDP and active GTP forms. The most common mutations in tumors lead to a loss of the ability to hydrolyze GTP. Thus, the Ras protein is trapped in the “on” position and continues to stimulate cell growth, even in the absence of a continuing signal.
Other genes can contribute to cancer development only when both copies of the gene normally present in a cell are deleted or otherwise damaged. Such genes are called tumor-suppressor genes. For example, genes for some of the phosphatases that participate in the termination of EGF signaling are tumor suppressors. Without any functional phosphatase present, EGF signaling persists once initiated, stimulating inappropriate cell growth.
Monoclonal antibodies can be used to inhibit signal-transduction pathways activated in tumors
Mutated or overexpressed receptor tyrosine kinases are frequently observed in tumors. For instance, the epidermal-growth-factor receptor (EGFR) is overexpressed in some human epithelial cancers, including breast, ovarian, and colorectal cancer. Because some small amount of the receptor can dimerize and activate the signaling pathway even without binding to EGF, overexpression of the receptor increases the likelihood that a “grow and divide” signal will be inappropriately sent to the cell. This understanding of cancer-related signal-transduction pathways has led to a therapeutic approach that targets the EGFR. The strategy is to produce monoclonal antibodies to the extracellular domains of the offending receptors. One such antibody, cetuximab (Erbitux), has effectively targeted the EGFR in colorectal cancers. Cetuximab inhibits the EGFR by competing with EGF for the binding site on the receptor. Because the antibody sterically blocks the change in conformation that exposes the dimerization arm, the antibody itself cannot induce dimerization. The result is that the EGFR-controlled pathway is not initiated.
Cetuximab is not the only monoclonal antibody that has been developed to target a receptor tyrosine kinase. Trastuzumab (Herceptin) inhibits another EGFR family member, HER2, that is overexpressed in approximately 30% of breast cancers. Recall that this protein can signal even in the absence of ligand, so it is especially likely that overexpression will stimulate cell proliferation. Breast-cancer patients are now being screened for HER2 overexpression and treated with Herceptin as appropriate. Thus, this cancer treatment is tailored to the genetic characteristics of the tumor.
Protein kinase inhibitors can be effective anticancer drugs
The widespread occurrence of overactive protein kinases in cancer cells suggests that molecules that inhibit these enzymes might act as antitumor agents. For example, more than 90% of patients with chronic myelogenous leukemia (CML) show a specific chromosomal defect in cancer cells (Figure 14.34). The translocation of genetic material between chromosomes 9 and 22 causes the c-abl gene, which encodes a tyrosine kinase of the Src family, to be inserted into the bcr gene on chromosome 22. The result is the production of a fusion protein called Bcr-Abl that consists primarily of sequences for the c-Abl kinase. However, the bcr-abl gene is not regulated appropriately; it is expressed at higher levels than that of the gene encoding the normal c-Abl kinase, stimulating a growth-promoting pathway. Because of this overexpression, leukemia cells express a unique target for chemotherapy. A specific inhibitor of the Bcr-Abl kinase, Gleevec (STI-571, imatinib mesylate), has proved to be a highly effective treatment for patients suffering from CML. This approach to cancer chemotherapy is fundamentally distinct from most approaches, which target all rapidly growing cells, including normal ones. Because Gleevec targets tumor cells specifically, side effects caused by the impairment of normal dividing cells can be minimized. Thus, our understanding of signal-transduction pathways is leading to conceptually new disease treatment strategies.
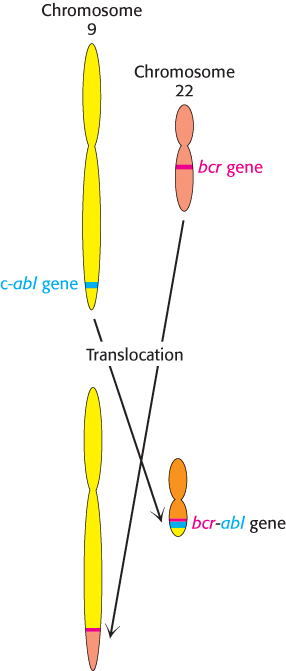
Figure 14.34: Formation of the bcr-abl gene by translocation. In chronic myelogenous leukemia, parts of chromosomes 9 and 22 are reciprocally exchanged, causing the bcr and abl genes to fuse. The protein kinase encoded by the bcr-abl gene is expressed at higher levels in tumor cells than is the c-abl gene in normal cells.
Cholera and whooping cough are the result of altered G-protein activity
Although defects in signal-transduction pathways have been most extensively studied in the context of cancer, such defects are important in many other diseases. Cholera and whooping cough are two pathologies of the G-protein-dependent signal pathways. Let us first consider the mechanism of action of the cholera toxin, secreted by the intestinal bacterium Vibrio cholerae. Cholera is a potentially life-threatening, acute diarrheal disease transmitted through contaminated water and food. It causes the voluminous secretion of electrolytes and fluids from the intestines of infected persons. The cholera toxin, also called choleragen, is a protein composed of two functional units—a β subunit that binds to GM1 gangliosides (Section 26.1) of the intestinal epithelium and a catalytic A subunit that enters the cell.
The A subunit catalyzes the covalent modification of a Gαs protein: the α subunit is modified by the attachment of an ADP-ribose to an arginine residue. This modification stabilizes the GTP-bound form of Gαs, trapping the molecule in its active conformation. The active G protein, in turn, continuously activates protein kinase A. PKA opens a chloride channel and inhibits sodium absorption by the Na+–H+ exchanger by phosphorylating both the channel and the exchanger. The net result of the phosphorylation is an excessive loss of NaCl and the loss of large amounts of water into the intestine. Patients suffering from cholera may pass as much as twice their body weight in fluid in 4 to 6 days. Treatment consists of rehydration with a glucose–electrolyte solution.
Whereas cholera is a result of a G protein trapped in the active conformation, causing the signal-transduction pathway to be persistently stimulated, pertussis is a result of the opposite situation. Pertussis toxin is secreted by Bordetella pertussis, the bacterium responsible for whooping cough. Like choleragen, pertussis toxin adds an ADP-ribose moiety to a Gα subunit. However, in this case, the ADP-ribose group is added to a Gαi protein, a Gα subunit that inhibits adenylate cyclase, closes Ca2+ channels, and opens K+ channels. The effect of this modification is to prevent binding of the heterotrimeric Gi protein to its receptor, trapping it in the “off” conformation. The pulmonary symptoms have not yet been traced to a particular target of the Gαi protein.