23.6Inborn Errors of Metabolism Can Disrupt Amino Acid Degradation
Inborn Errors of Metabolism Can Disrupt Amino Acid Degradation
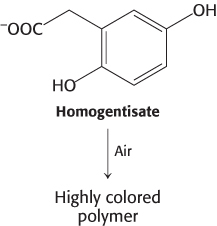
 Errors in amino acid metabolism provided some of the first examples of biochemical defects linked to pathological conditions. For instance, alcaptonuria, an inherited metabolic disorder caused by the absence of homogentisate oxidase, was described in 1902. Homogentisate, a normal intermediate in the degradation of phenylalanine and tyrosine (Figure 23.27), accumulates in alcaptonuria because its degradation is blocked. Homogentisate is excreted in the urine, which turns dark on standing as homogentisate is oxidized and polymerized to a melanin-
Errors in amino acid metabolism provided some of the first examples of biochemical defects linked to pathological conditions. For instance, alcaptonuria, an inherited metabolic disorder caused by the absence of homogentisate oxidase, was described in 1902. Homogentisate, a normal intermediate in the degradation of phenylalanine and tyrosine (Figure 23.27), accumulates in alcaptonuria because its degradation is blocked. Homogentisate is excreted in the urine, which turns dark on standing as homogentisate is oxidized and polymerized to a melanin-
Although alcaptonuria is a relatively harmless condition, such is not the case with other errors in amino acid metabolism. In maple syrup urine disease, the oxidative decarboxylation of α-ketoacids derived from valine, isoleucine, and leucine is blocked because the branched-
|
Disease |
Enzyme deficiency |
Symptoms |
|---|---|---|
|
Citrullinema |
Arginosuccinate lyase |
Lethargy, seizures, reduced muscle tension |
|
Tyrosinemia |
Various enzymes of tyrosine degradation |
Weakness, liver damage, mental retardation |
|
Albinism |
Tyrosinase |
Absence of pigmentation |
|
Homocystinuria |
Cystathionine β-synthase |
Scoliosis, muscle weakness, mental retardation, thin blond hair |
|
Hyperlysinemia |
α-Aminoadipic semialdehyde dehydrogenase |
Seizures, mental retardation, lack of muscle tone, ataxia |
706
Phenylketonuria is one of the most common metabolic disorders
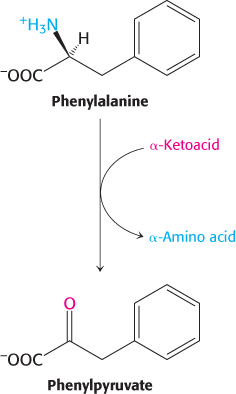
Phenylketonuria is perhaps the best known of the diseases of amino acid metabolism. Phenylketonuria, which occurs with a prevalence of 1 in 10,000 births, is caused by an absence or deficiency of phenylalanine hydroxylase or, more rarely, of its tetrahydrobiopterin cofactor. Phenylalanine accumulates in all body fluids because it cannot be converted into tyrosine. Normally, three-
Almost all untreated phenylketonurics are severely mentally retarded. The brain weight of these people is below normal, myelination of their nerves is defective, and their reflexes are hyperactive. The life expectancy of untreated phenylketonurics is drastically shortened. Half die by age 20 and three-
Early diagnosis of phenylketonuria is essential and has been accomplished by mass screening programs of all babies born in the United States and Canada. The phenylalanine level in the blood is the preferred diagnostic criterion because it is more sensitive and reliable than the FeCl3 test. Prenatal diagnosis of phenylketonuria with DNA probes has become feasible because the gene has been cloned and the exact locations of many mutations have been discovered in the protein.
Determining the basis of the neurological symptoms of phenylketonuria is an active area of research
The biochemical basis of retardation is not firmly established, but one hypothesis suggests that the lack of hydroxylase reduces the amount of tyrosine, an important precursor to neurotransmitters such as dopamine. Moreover, high concentrations of phenylalanine prevent amino acid transport of any tyrosine present as well as tryptophan, a precursor to the neurotransmitter serotonin, into the brain. Because all three of the amino acids are transported by the same carrier, phenylalanine will saturate the carrier, preventing access to tyrosine and tryptophan. Finally, high blood levels of phenylalanine result in higher levels of phenylalanine in the brain, and evidence suggests this elevated concentration inhibits glycolysis at pyruvate kinase, disrupts myelination of nerve fibers and reduces the synthesis of several neurotransmitters.
707