25.5Disruptions in Nucleotide Metabolism Can Cause Pathological Conditions
Disruptions in Nucleotide Metabolism Can Cause Pathological Conditions
Nucleotides are vital to a host of biochemical processes. It is not surprising, then, that disruption of nucleotide metabolism would have a variety of physiological effects. The nucleotides of a cell undergo continual turnover. Nucleotides are hydrolytically degraded to nucleosides by nucleotidases. The phosphorolytic cleavage of nucleosides to free bases and ribose 1-
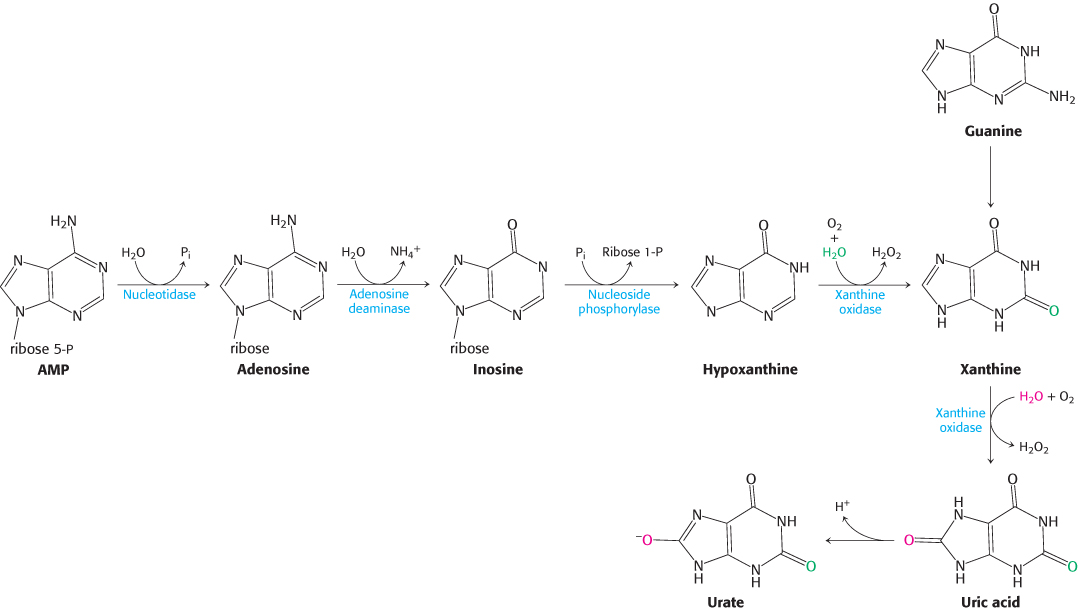
The loss of adenosine deaminase activity results in severe combined immunodeficiency
 The pathway for the degradation of AMP includes an extra step because adenosine is not a substrate for nucleoside phosphorylase. First, the phosphate is removed by a nucleotidase to yield the nucleoside adenosine (Figure 25.17). In the extra step, adenosine is deaminated by adenosine deaminase to form inosine.
The pathway for the degradation of AMP includes an extra step because adenosine is not a substrate for nucleoside phosphorylase. First, the phosphate is removed by a nucleotidase to yield the nucleoside adenosine (Figure 25.17). In the extra step, adenosine is deaminated by adenosine deaminase to form inosine.
A deficiency in adenosine deaminase activity is associated with some forms of severe combined immunodeficiency (SCID), an immunological disorder. Persons with the disorder have acute recurring infections, often leading to death at an early age. SCID is characterized by a loss of T cells, which are crucial to the immune response (Section 34.5). Although the biochemical basis of the disorder is not clearly established, a lack of adenosine deaminase results in an increase of 50 to 100 times the normal level of dATP, which inhibits ribonucleotide reductase and, consequently, DNA synthesis. Moreover, adenosine itself is a powerful signal molecule with a role in a number of regulatory pathways. Disruption in the levels of adenosine may also be deleterious. SCID is often called the “bubble boy disease” because its treatment may include complete isolation of the patient from the environment. Adenosine deaminase deficiency has been successfully treated by gene therapy.
761
Gout is induced by high serum levels of urate
Inosine generated by adenosine deaminase is subsequently metabolized by nucleoside phosphorylase to hypoxanthine. Xanthine oxidase, a molybdenum-
High serum levels of urate (hyperuricemia) induce the painful joint disease gout. In this disease, the sodium salt of urate crystallizes in the fluid and lining of the joints. The small joint at the base of the big toe is a common site for sodium urate buildup, although the salt accumulates at other joints also. Painful inflammation results when cells of the immune system engulf the sodium urate crystals. The kidneys, too, may be damaged by the deposition of urate crystals. Gout is a common medical problem, affecting 1% of the population of Western countries. It is nine times as common in men as in women.
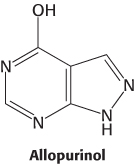
Administration of allopurinol, an analog of hypoxanthine, is one treatment for gout. The mechanism of action of allopurinol is interesting: it acts first as a substrate and then as an inhibitor of xanthine oxidase. The oxidase hydroxylates allopurinol to alloxanthine (oxipurinol), which then remains tightly bound to the active site. The binding of alloxanthine keeps the molybdenum atom of xanthine oxidase in the +4 oxidation state instead of it returning to the +6 oxidation state as in a normal catalytic cycle. We see here another example of suicide inhibition.
The synthesis of urate from hypoxanthine and xanthine decreases soon after the administration of allopurinol. The serum concentrations of hypoxanthine and xanthine rise, and that of urate drops.
 The average serum level of urate in human beings is close to the solubility limit and is higher than levels found in other primates. What is the selective advantage of a urate level so high that it teeters on the brink of gout in many people? It turns out that urate has a markedly beneficial action. Urate is a highly effective scavenger of reactive oxygen species. Indeed, urate is about as effective as ascorbate (vitamin C) as an antioxidant. The increased level of urate in human beings may protect against reactive oxygen species that are implicated in a host of pathological conditions (Table 18.3).
The average serum level of urate in human beings is close to the solubility limit and is higher than levels found in other primates. What is the selective advantage of a urate level so high that it teeters on the brink of gout in many people? It turns out that urate has a markedly beneficial action. Urate is a highly effective scavenger of reactive oxygen species. Indeed, urate is about as effective as ascorbate (vitamin C) as an antioxidant. The increased level of urate in human beings may protect against reactive oxygen species that are implicated in a host of pathological conditions (Table 18.3).
Lesch–Nyhan syndrome is a dramatic consequence of mutations in a salvage-pathway enzyme
Mutations in genes that encode nucleotide biosynthetic enzymes can reduce levels of needed nucleotides and can lead to an accumulation of intermediates. A nearly total absence of hypoxanthine-
762
What is the connection between the absence of HGPRT activity and the behavioral characteristics of Lesch-
Folic acid deficiency promotes birth defects such as spina bifida
Spina bifida is one of a class of birth defects characterized by the incomplete or incorrect formation of the neural tube early in development. In the United States, the prevalence of neural-