4.2 SECONDARY STRUCTURE
Secondary structure refers to regularly repeating elements within a protein, in which hydrogen bonds form between polar atoms in the backbone chain. These hydrogen-bonded structures shield, or neutralize, many charges and allow the intrinsically polar polypeptide chain to traverse the nonpolar interior of a protein. The main secondary structures are the α helix, typically 10 to 15 residues long, and the β conformation, composed of individual segments (called β strands) of 3 to 10 residues. A typical protein contains about one-third α helix and one-third β conformation, although there are plenty of exceptions to this general rule, including proteins that have only one of these types of secondary structure. The portion of a protein that has neither α helices nor β conformation is composed of loops and turns that allow secondary structural elements to reverse direction back and forth to form a folded, globular protein. Here we describe the structure and properties of α helices and the β conformation and briefly discuss the structure of reverse turns, which allow secondary structures to fold.
The α Helix Is a Common Form of Protein Secondary Structure
The α helix was originally predicted by Pauling and Corey in 1951, based on x-ray studies of keratin by William Astbury in the 1930s. The α helix contains 3.6 amino acid residues per turn (Figure 4-6a). One full turn of the α helix is 5.4 Å (1.5 Å per residue) long, and the R groups protrude outward from the helix. The hydrogen on the amide nitrogen forms a hydrogen bond with the carbonyl oxygen of the fourth residue toward the N-terminus, which makes about one helical turn. The α helix forms a right-handed spiral, which, moving away from an observer looking down the spiral, corresponds to a clockwise rotation. You can determine the chirality of a spiral (i.e., whether right- or left-handed) using your hands (Figure 4-6b). With your fingers making a fist and your thumbs sticking out and pointing away from you, a left-handed spiral would appear to curve in the same direction as the fingers on your left hand, in a counterclockwise rotation, as the spiral projects in the direction of your thumb. A right-handed spiral, such as the α helix, curves in the same direction as the fingers of your right hand, as the spiral projects in the direction of your right thumb.
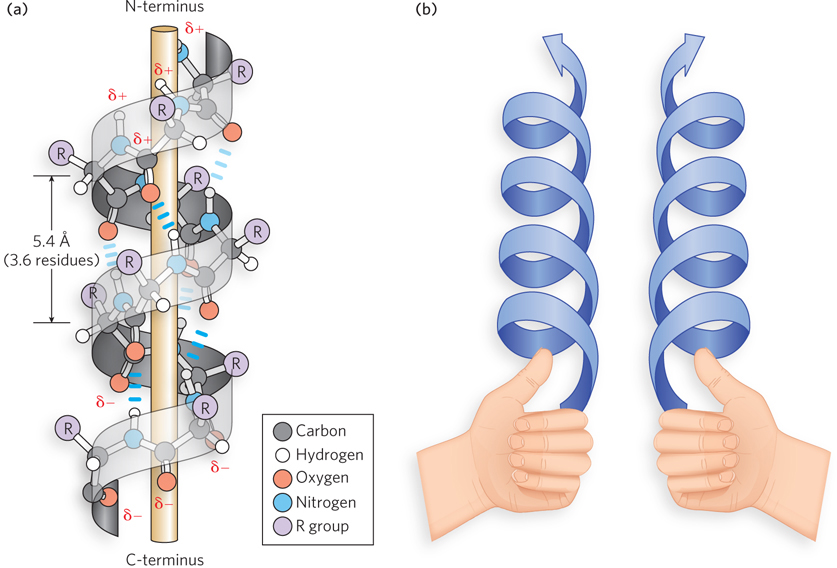
Figure 4-6: The structure of the α helix. (a) Peptide bonds form a right-handed spiral; intrachain hydrogen bonds are shown. The electric dipole of the helix, established through intrachain hydrogen bonds, propagates to the amino and carbonyl constituents of each peptide bond. The partial charges of the electric dipole are indicated by δ+ and δ−. (b) An easy way to distinguish left- and right-handed helices (see text).
All the hydrogen bonds of an α helix point in the same direction, and this sets up an electric dipole that gives a partial positive charge to the N-terminus and a partial negative charge to the C-terminus of the helix. Because the last four residues at either end of an α helix are not fully hydrogen-bonded, the dipole charges are spread out on these residues. For this reason, the conformations at the ends of an α helix are often irregular or form a more strained version of the α helix with less favorable torsion angles.
Some general guidelines enable us to predict from a protein sequence the sections where an α helix will form. Consecutive stretches of amino acid residues with long or bulky R groups cannot approach one another closely enough to form the tightly packed α helix. Also, polar side chains can hydrogen-bond to the peptide backbone, thereby destabilizing the helix. For this reason, serine, asparagine, aspartate, and threonine are found less frequently in α helices than most other amino acids. In addition, consecutive like-charged R groups repel one another in the close confines of the α helix. Glycine, due to its conformational flexibility, is also infrequently found in α helices. Finally, proline is infrequent in α helices because its cyclic structure lacks an amide hydrogen-bond donor and restricts N–Cα bond rotation. Proline is often referred to as a helix-breaking residue. The relative frequency of the 20 amino acids in different types of secondary structure is shown in Figure 4-7.
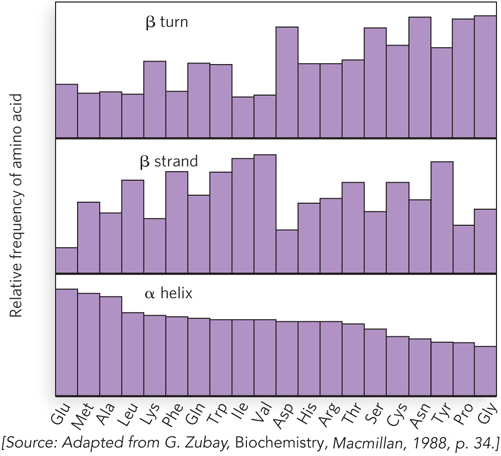
Figure 4-7: The relative frequency of amino acids in secondary structural elements. The plot shows the observed relative frequencies of the 20 common amino acids in three types of secondary structure.
Some arrangements of amino acid residues can stabilize the helix. For example, side chains spaced four residues apart are stacked upon one another in the helix. Oppositely charged side chains that are close together can form an ion pair, which stabilizes the helix. Likewise, aromatic side chains with this four-residue spacing can form hydrophobic effects that stabilize the helix. Amino acids with a charge opposite to the partial charge of the helix dipole are sometimes located at the ends of a helix, which adds stability.
The β Conformation Forms Sheetlike Structures
A segment of peptide in the β conformation rarely occurs alone. Instead, multiple segments of β conformation are arranged side by side to form a β sheet consisting of at least two, and frequently many more, β strands (Figure 4-8). The β sheet, like the α helix, is formed by hydrogen bonds between backbone amide and carbonyl groups, but unlike the α helix, the β sheet structure cannot form from one β strand. Instead, all hydrogen bonds are formed between the backbones of two different β strands. The peptide bonds in a β sheet are arranged in a remarkably extended form, with a distance of 3.5 Å per residue. The R groups of adjacent amino acid residues in a β strand lie on opposite sides of the sheet, and this alternating geometry prevents the interaction of R groups of adjacent residues. This sets up a zigzag pattern and, along with the side-to-side arrangement of the strand segments, resembles a series of pleats. Thus, the β sheet is often referred to as a “β-pleated sheet.”
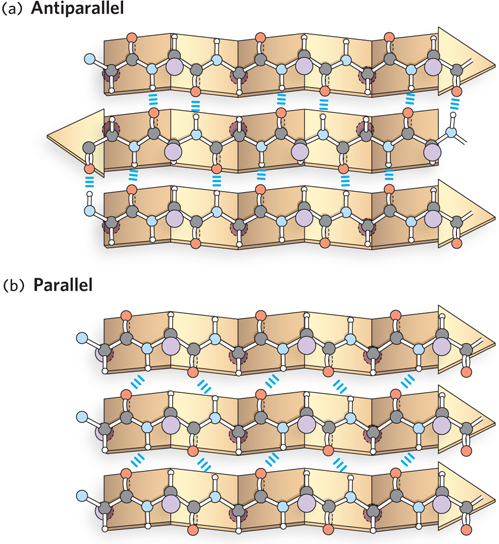
Figure 4-8: The structure of the β sheet. R groups extend out from the β sheet, emphasizing the pleated shape. Hydrogen bonds between adjacent β strands are also shown. (a) In an antiparallel β sheet, the N- to C-terminal orientation of the β strands alternates (shown by the arrowheads). (b) In a parallel β sheet, the β strands align in the same direction.
Often, the many β segments or strands that compose a β sheet are covalently connected in a single polypeptide. The strands of a β sheet may be close together in the polypeptide sequence, but they can also be far apart, separated by other secondary structures in the same polypeptide chain. The formation of β sheets between two different polypeptides is also common. In either case, when the β strands are oriented in the opposite N- to C-terminal directions, the structure is known as an antiparallel β sheet; when they run in the same direction, it is called a parallel β sheet (see Figure 4-8). The sheets can also be composed of a mixture of parallel and antiparallel strands. Sheets made with antiparallel strands form nearly straight hydrogen bonds and are thought to be slightly more stable than parallel sheet structures. The β sheet structure can readily accommodate large aromatic residues, such as Tyr, Trp, and Phe residues. In addition, proline, which is unfavored in the α helix, is often found in β sheets, especially in the “edge” strands, perhaps to prevent association between proteins that are not meant to bind one another. The most common sheet structures are antiparallel, followed by mixed sheets, then purely parallel sheets. Because alternating R groups in β sheets are on opposite sides of the sheet structure, hydrophobic residues that alternate with polar side chains can yield a sheet structure that acts as a boundary between greasy and watery environments.
Reverse Turns Allow Secondary Structures to Fold
The size of α helices and β strands is limited by the diameter of a globular protein, and these structures must repeatedly reverse direction back and forth to form a properly folded protein. Approximately one-third of a polypeptide chain is composed of reverse turns, or loops, where secondary structural elements reverse themselves. Sometimes these turns are large and irregular, but many reverse turns are small and precise; these are called β turns (Figure 4-9a). The β turn makes a complete reversal of direction using only four residues, in which the backbone carbonyl oxygen of the first residue forms a hydrogen bond with the amide hydrogen of the fourth residue. The second and third residues form no inter-residue hydrogen bonds, and this typically places β turns on the surface of the protein, where the backbone can hydrogen-bond to water.
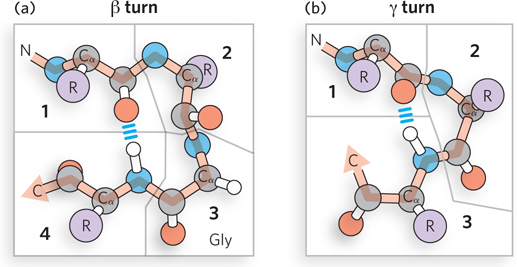
Figure 4-9: The linking of secondary structural elements by reverse turns. (a) An example of the β turn, involving four amino acid residues. (b) The much less common γ turn, accomplished by three residues.
There are eight types of β turns, classified according to the torsion angles between the inner two amino acid residues. Often, β turns contain a Pro residue at position 2 and Gly residue at position 3. Glycine is prevalent in β turns because its R group (hydrogen) allows it to accommodate many conformations that are not possible for other amino acids (see Figure 4-3). Although the conformation of a Pro residue is highly restricted compared with other amino acids, the imino nitrogen of proline can readily assume a cis configuration (see Figure 4-4b), a form that is especially suitable for tight turns.
A much less common reverse turn is the γ turn, consisting of only three amino acids (Figure 4-9b). The backbone carbonyl and amide groups of the first and third amino acid residues form a hydrogen bond, and the second (middle) amino acid of the γ turn is not involved in inter-residue hydrogen bonding.
SECTION 4.2 SUMMARY
Secondary structure is a regularly repeating element that has hydrogen bonds between atoms of the peptide backbone.
The α helix contains 3.6 amino acid residues per turn, and its internal hydrogen bonds set up an electric dipole for the entire helix.
The compact structure of the α helix is unfavorable for certain combinations of residues, such as consecutive residues that are like-charged or bulky; the confined geometry of the proline ring can distort the helix, and proline is sometimes referred to as a helix breaker.
The β sheet is formed by hydrogen bonding between two peptide segments in the β conformation, either within a polypeptide chain or between separate polypeptide chains. It can have a parallel or antiparallel configuration.
The extended structure of a β sheet can accommodate most amino acid residues, including bulky aromatic amino acids and proline.
Secondary structural elements in a polypeptide are connected by reverse turns. These may consist of long, unstructured stretches of amino acid residues or tighter, precise structures consisting of four amino acids (β turns) or, more rarely, three amino acids (γ turns).