Chapter 4
Question 4.1
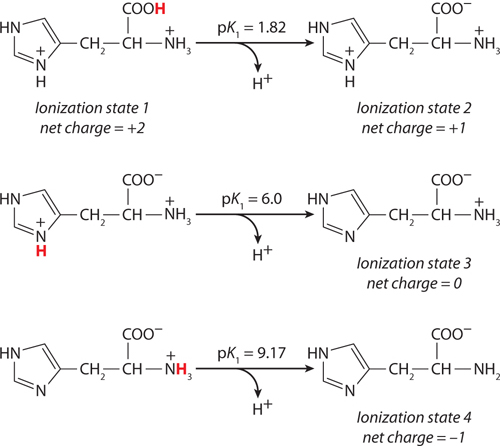
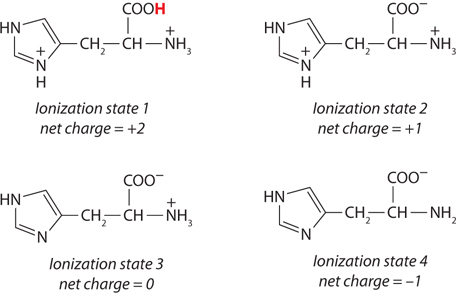
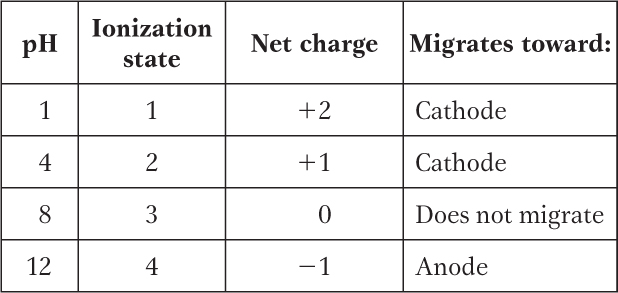
Question 4.2
(a) Minimum Mr 32,000. Remember that the molecular weight of a Trp residue is not the same as the molecular weight of the free amino acid. (b) 2 Trp residues.
Question 4.3
(a) At pH 3, +2; at pH 8, 0; at pH 11, −1. (b) pI 8.
Details:
This peptide has five ionizable groups: (1) the α-amino group of E (pKa 9.67); (2) the side chain of E (pKa 4.25); (3) the side chain of H (pKa 6.0); (4) the side chain of R (pKa 12.48); and (5) the α-carboxyl group of G (pKa 2.34). At pH 3, (1), (3), and (4) are protonated and positively charged, giving a net charge for these groups of +3. The pH of 3 is between the two pKa values for (2) and (5) (one mostly protonated, the other mostly unprotonated), giving a net charge of about −1 for these two groups. This yields a net charge for the peptide of about +2. At pH 8, (2) and (5) are unprotonated, contributing a charge of −2; (3) is mostly unprotonated and neutral; (1) and (4) are mostly protonated, contributing a charge of nearly +2. The net charge is near zero. At pH 11, (2), (3), and (5) are unprotonated, with a charge of −2; (1) is mostly unprotonated and uncharged, while (4) is mostly protonated, contributing a net charge of +1. The net charge is close to −1.
The pI can be estimated by determining the pH at which the net charge is zero. In this peptide, (1), (3), and (4) can contribute + charges when protonated, and (2) and (5) can contribute − charges when unprotonated. Hence, a net charge of zero can occur at the pH where (1), (3), and (4) together contribute a net +2 charge to exactly balance the net −2 contributed by the two carboxyl groups of (2) and (5). This will occur about halfway between the pKa values of the H and R side chain and the α-amino group of E: here, the fraction of E α-amino groups that are unprotonated is balanced exactly by the fraction of H side chains that are protonated. Thus, the pI is approximately 8.
S-
Question 4.4
(a) A1, R5, K20, R28. (b) D2, E4, D27, E29, T30. (c) C7, C23.
Question 4.5
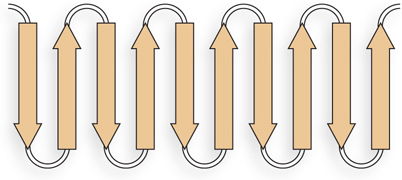
The sheets are likely to be antiparallel, because the linkers that connect one β strand to the next are too short to connect parallel strands. The linkers are four residues long and could form β turns.
Question 4.6
Alternating R groups are on opposite sides of the β sheet structure. Therefore, in a continuous layer of β sheet, with alternating polar and nonpolar residues in the β strands, the sheet is likely to fold into a β barrel, sequestering hydrophobic residues inside.
Question 4.7
Residues 1 and 3 are in β sheets, residue 2 is in a right-
Question 4.8
 The P3 and P18 residues disfavor helix formation, limiting the region favorable for helix formation to the sequence between residues 4 and 17. The several positively charged residues, 4 through 7, are not likely to initiate a helix, because they would repel one another and interact unfavorably with the helix dipole, which is positively charged at the N-
The P3 and P18 residues disfavor helix formation, limiting the region favorable for helix formation to the sequence between residues 4 and 17. The several positively charged residues, 4 through 7, are not likely to initiate a helix, because they would repel one another and interact unfavorably with the helix dipole, which is positively charged at the N-
Question 4.9
(a) and (d). Both contain the consensus sequence for an ATP/GTP-
Question 4.10
As an α helix, 210 Å ([1.5 Å/residue] × 140 residues). As a β strand, 490 Å ([3.5 Å/residue] × 140 residues).
Question 4.11
There are many possible answers; any of the following will suffice. NMR uses magnets and radiofrequency irradiation; crystallography uses x rays. NMR is performed on proteins in solution; x-
Question 4.12
(a), (b), (c) one domain; (d), (e) two domains. When a protein reaches a size of about 150 to 200 residues (Mr ∼20,000), the polypeptide chain usually folds into two domains.
Question 4.13
Completely buried residues are likely to be hydrophobic: L2, F4, I6, V8, V12, L13, L18, and L19 fit this description. Highly polar residues, or at least their polar groups, are likely to be on the surface, exposed to water: D1, K3, T5, S7, T14, R15, E16, Q17, and E20 fit this description.
Question 4.14
(a) Destabilizes the positive dipole at the N-
Question 4.15
(a) The N-
Question 4.16
Question 4.17
(a) Bovine serum albumin. (b) Green fluorescent protein.
Details: Much of the structure of bovine serum albumin is in the form of an α helix, while green fluorescent protein is predominantly in the β conformation. The corresponding Ramachandran plots exhibit many residues with torsion angles characteristic of the predominant secondary structures.
Question 4.18
The conversion of trans peptide bonds to cis peptide bonds is catalyzed by an enzyme, peptide prolyl cis-
Question 4.19
No correct answer is possible at this stage of the book. However, the answer is yes: virtually all of these complexes, no matter how large, are held together by weak noncovalent interactions.