14.3 THE EVOLUTIONARY INTERPLAY OF TRANSPOSONS AND THEIR HOSTS
As we noted in the introduction to this chapter, transposition provides an alternative survival strategy that almost certainly has ancient roots. Evolution has given rise to many types of transposons, which have colonized the genomes of all extant organisms, and it has also given rise to other, more complex pathogens, such as viruses. Although the need to adapt to their hosts has affected transposon evolution, transposons have not always remained separate entities. Some transposable elements, and the enzymes they encode, have been appropriated by host cells and adapted to new biological tasks. The movements of transposons have driven genomic changes that have contributed in important ways to evolution.
Viruses, Transposons, and Introns Have an Interwoven Evolutionary History
Many well-
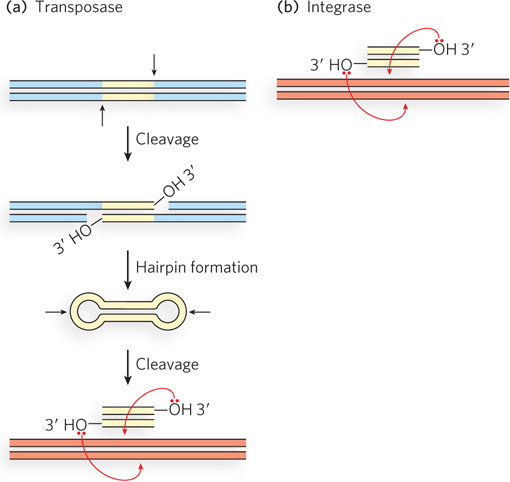
The evolution of virtually all retrotransposons and retroviruses can be traced through their reverse transcriptase genes. Similarly, the evolution of an even broader range of transposable elements can be linked to the evolution of retrotransposons and retroviruses through their integrase and transposase genes. Integrases and transposases catalyze very similar reactions (Figure 14-20). The most widespread class of both types of enzymes uses a set of three active-
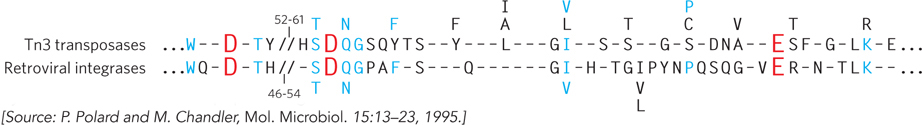
Transposons have a long and complex history. Their dispersal may include rare events in which DNA was transferred by some means (such as bacterial conjugation, cellular fusion, viral infection, or accidental DNA uptake) among the cells of different species. When a transposon is introduced into a new species’ genome, there is often a period of many host generations during which the element transposes more or less freely. The number of inserted transposons may increase, with the resulting genomic changes being passed on whenever they do not have a deleterious effect on the host. As time passes, the transposons become subject to silencing processes, including the introduction of mutations in their transposase or integrase genes that inactivate the gene products. Alternatively, the host may find a way to shut down transposition. One common silencing mechanism involves RNA interference (RNAi, a process described in Chapter 22). In brief, the cell produces short RNA molecules that are complementary to the transcripts of the transposase-
509
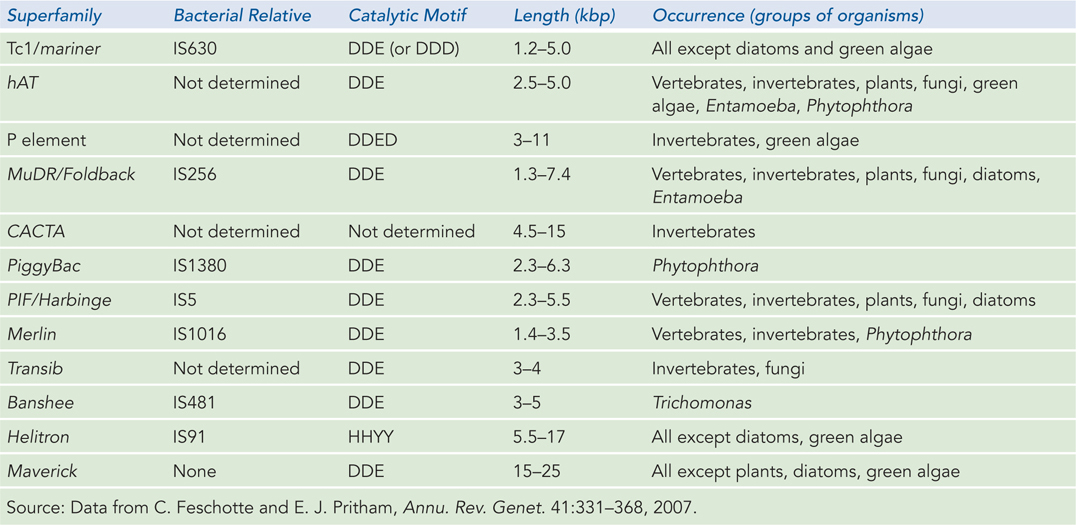
Linear transmission of transposons from one host generation to the next is predominant, with transfer between species occurring rarely. Thus, many transposon families are found only in certain classes of organisms. In eukaryotes, ongoing genomic sequencing efforts have revealed 12 superfamilies of DNA transposons, including Tc1/mariner (Table 14-1). Many of these superfamilies are found in more than one eukaryotic type. Seven are closely related to transposons found in bacteria, suggesting that they appeared before the divergence of bacteria and eukaryotes.
Sometimes transposons benefit their hosts. As we have seen, the antibiotic-
Perhaps more important is the overall impact of transposons on the evolution of the host. Genomic changes promoted by transposons come in many forms. Transposons are set up to bring their ends together in a complex prior to any cleavage event, but this control mechanism can go awry. If transposase subunits form a complex involving two ends derived from different copies of the same transposon, on the same or different chromosomes, large genomic rearrangements can result. Genes may be captured between two transposable elements and moved to different genomic locations. If the genes are duplicated in the process, the new gene copies may evolve and acquire new functions. Transposition is not always precise; the insertion of a transposon into a gene, followed by its later excision, can add or subtract base pairs in the gene and create new alleles. Also, the insertion or excision of transposons at particular genomic sites can alter the expression of genes or sets of genes.
510
The transposons that seem to clutter mammalian genomes have been referred to as “selfish” or “junk” DNA, but these labels are being shed as our understanding broadens. Transposon DNA may play a key role in chromosomal structure and packaging. And far from being dormant, transposon DNA is actively transcribed in at least some cells. As new classes of functional RNAs are being discovered at a rapid pace, the RNAs produced by transposons may prove to have unexpected cellular roles.
A Hybrid Recombination Process Assembles Immunoglobulin Genes
Humans have a complex immune system capable of generating millions of different immunoglobulins (antibody proteins) with distinct binding specificities. But the human genome contains only about 25,000 genes, and just a few hundred of these are immune system genes. Somehow, the millions of different immunoglobulins are generated from these several hundred genes. As B lymphocytes (B cells) differentiate, their immunoglobulin genes recombine so that each cell will express an antibody with a unique binding specificity. Studies of the recombination mechanism reveal a close relationship to DNA transposition and suggest that this system for generating antibody diversity evolved from an ancient cellular invasion by transposons.
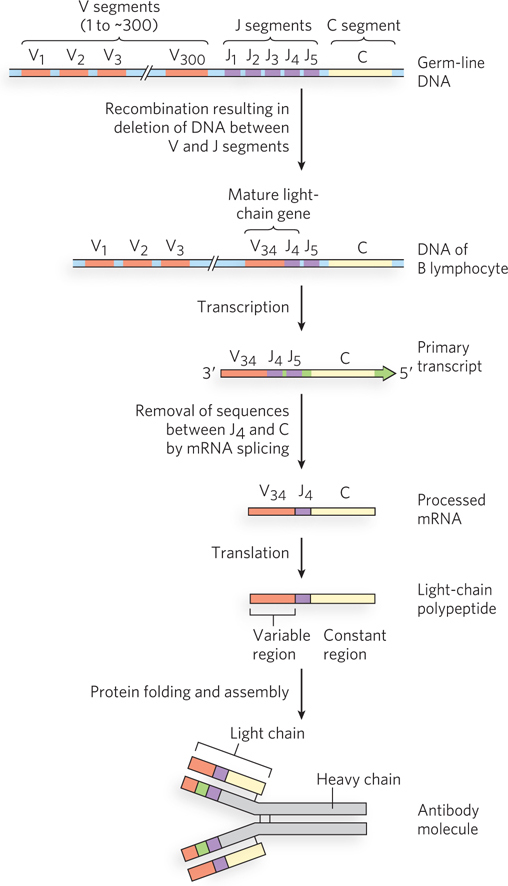
Immunoglobulins consist of two heavy and two light polypeptide chains (Figure 14-22 shows the general structure of the IgG class of immunoglobulins). Each chain has two regions: a variable region, with a sequence that differs greatly from one immunoglobulin to another, and a constant region, which is virtually unchanging within a class of immunoglobulins. There are two distinct families of light chains, kappa and lambda, which differ somewhat in the sequence of their constant regions. For all three types of polypeptide chain (heavy chain, and kappa and lambda light chains), diversity in the variable regions is generated by a similar mechanism. The genes for these polypeptides are divided into segments, and the genome contains clusters with multiple versions of each segment. The joining of one version of each gene segment creates a complete gene.
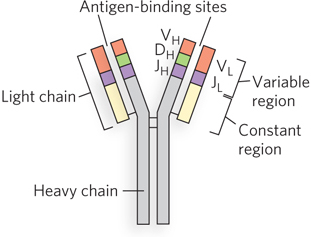
Figure 14-23 depicts the organization of the DNA encoding the kappa light chain and shows how a mature kappa light chain is generated. In undifferentiated cells, the coding information for this polypeptide is separated into three segments. The V (variable) segment encodes the first 95 amino acid residues of the variable region, the J (joining) segment encodes the remaining 12 residues of the variable region, and the C (constant) segment encodes the constant region. For kappa light chains, the genome contains ∽300 different V segments, 5 different J segments, and 1 type of C segment.
511
As a stem cell in the bone marrow differentiates to form a mature B cell, one V segment and one J segment are brought together by a specialized recombination system (Figure 14-24). During this programmed DNA deletion, the intervening DNA is discarded. There are about 300 × 5 = 1,500 possible V-
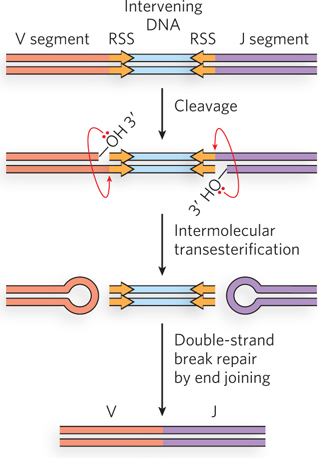
The recombination mechanism for joining the V and J segments is facilitated by recombination signal sequences (RSS) that lie just downstream of each V segment and just before each J segment. These sequences are bound by proteins called RAG1 and RAG2 (products of the recombination activating gene). The RAG proteins catalyze the formation of a double-
The genes for the heavy chains and the lambda light chains form by similar processes. Heavy chains have more gene segments than light chains, with thousands of possible combinations. Because any heavy chain can combine with any light chain to generate an immunoglobulin, each human can produce at least 107 possible immunoglobulins. And additional diversity is generated by high mutation rates (of unknown mechanism) in the V segments during B-
The mechanism of the DNA recombination events required to generate an expressed immunoglobulin gene suggests that the immune system evolved, in part, from ancient transposons. The mechanism for generation of the double-
SECTION 14.3 SUMMARY
Transposons, retrotransposons, and retroviruses have a shared evolutionary history, evident in the phylogenies of the key enzymes—
reverse transcriptases, transposases, and integrases— that promote these processes. 512
Important elements of the vertebrate immune system, the enzymes that promote immunoglobulin gene rearrangements and thus immunoglobulin diversity, evolved from the transposase/integrase family of enzymes. RAG1 is related to the transposases of the Transib transposons.
UNANSWERED QUESTIONS
For any organism, the information required for creating a new generation is passed on through its DNA. Stable transmission is needed, yet genomes are surprisingly dynamic. Recombination processes contribute to repair and facilitate key steps of replication and cell division. A hidden world of transposons makes a home in each genome, replicating passively yet contributing to evolution in important ways. This dynamic genome still holds some secrets to unlock.
What is the origin of reverse transcriptase? How does it relate to the origin of retroviruses? What is its impact on genome development and diversity? For researchers interested in the origin of life, reverse transcriptase is potentially a very old enzyme that played a key role in the transition from RNA-
to DNA- based life. Why do the types of transposons vary so much from one class of organism to another? Each class of transposon present in a given genome represents an invasion that occurred sometime in the lineage of that organism. The study of genomic transposons may provide a rich harvest of information about the evolutionary past of all organisms.
How many proteins and other factors are involved in controlling transposition? Exploration of the elaborate interface between transposons and their hosts is only just beginning. The processes that silence a transposon often involve genes found in both the transposon and the host. The extent of host gene involvement has not been fully explored in most cases; functional RNA molecules may do part of the work. Similarly, elaborate processes that prevent integration of transposons into other transposons are only partially understood. AIDS is, as yet, an almost intractable disease, in part because of the capacity of HIV to integrate into a host genome and remain there, replicating passively. A permanent cure for HIV cannot occur as long as these silent HIV genomes provide a potential source of new infection. A better understanding of how this integration is regulated may eventually lead to genomic clean-
up therapies to eliminate or permanently inactivate these pathogens. That understanding must come from work on a wide range of viruses and transposons to fully sample the variety of mechanisms they use, as well as to unearth host functions that play subtle roles. What do retroviruses and transposons contribute to their hosts? The evolutionary history of these pathogens is clearly not entirely shaped by their own requirements. Obvious contributions to host survival have already been described, but the sheer bulk of transposon DNA in the human genome inspires new questions about function. How do all these repeated transposon sequences affect the structure and function of chromosomes? New reports suggest that much of the genomic DNA previously labeled as junk is in fact transcribed. What are all these RNA molecules doing? For example, a newly discovered class of RNAs called piwi RNAs (piRNAs) are abundant in germ-
line cells (especially during spermatogenesis). They play a role in the silencing of transposon genes, but their origin and detailed function are still a mystery.
513
HOW WE KNOW: Bacteriophage λ Provided the First Example of Site-Specific Recombination
Echols, H. 2001. Operators and Promoters: The Story of Molecular Biology and Its Creators. Berkeley: University of California Press.
Gottesman, M.E., and R.A. Weisberg. 2004. Little lambda, who made thee? Microbiol. Mol. Biol. Rev. 68:796–
Nash, H.A. 1975. Integrative recombination of bacteriophage lambda DNA in vitro. Proc. Natl. Acad. Sci. USA 72:1072–

Since the 1950s, scientists have known that the DNA of bacteriophage λ (λ phage) is linked to its bacterial host chromosome at a specific chromosomal location. The correct explanation for how the λ DNA enters the chromosome appeared in 1962, before anyone knew that the linear λ DNA is circularized on entering a bacterial cell. Allan Campbell, then at the University of Rochester, had the novel insight that circularization, followed by recombination into the host chromosome, could explain many observations associated with λ lysogeny. Clearly, a uniquely precise recombination process was at work, one that used defined DNA sequences.
A molecular understanding of this, as yet unprecedented, reaction mechanism required an in vitro system—
In his in vitro system, Nash constructed an altered bacteriophage λ chromosome that included both recombination sites, by then defined and named attB and attP (B for bacterium and P for phage), separated by about 15% of the chromosome’s length (Figure 1). As a source of the required enzymes, Nash used a concentrated extract derived from cells in which λ proteins were being produced. He then showed that integrative recombination between the two recombination sites would occur in cells to produce phage chromosomes 15% smaller than normal.
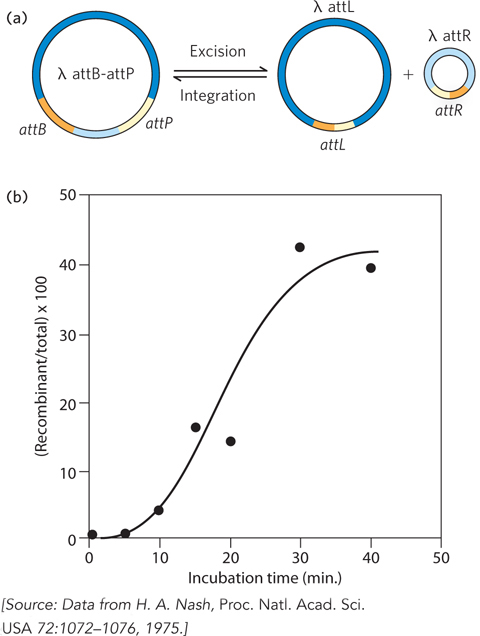
Phage with the shortened chromosome had the useful property that they were infectious in the presence of metal-
A successful in vitro system is a powerful thing in molecular biology. Nash, soon joined at the NIH by Kiyoshi Mizuuchi, used his system to purify the λ Int protein, discover a required host protein (IHF, for integration host factor), and define the reaction requirements. Following further work in other labs, notably that of Art Landy at Brown University, the λ integration system gradually yielded its secrets and stimulated the search for other site-
514
If You Leave out the Polyvinyl Alcohol, Transposition Gets Stuck
Craigie, R., and K. Mizuuchi. 1985. Mechanism of transposition of bacteriophage Mu: Structure of a transposition intermediate. Cell 41:867–

Understanding how a process works often starts with focusing on the reactions that are most efficient and easiest to detect and study—
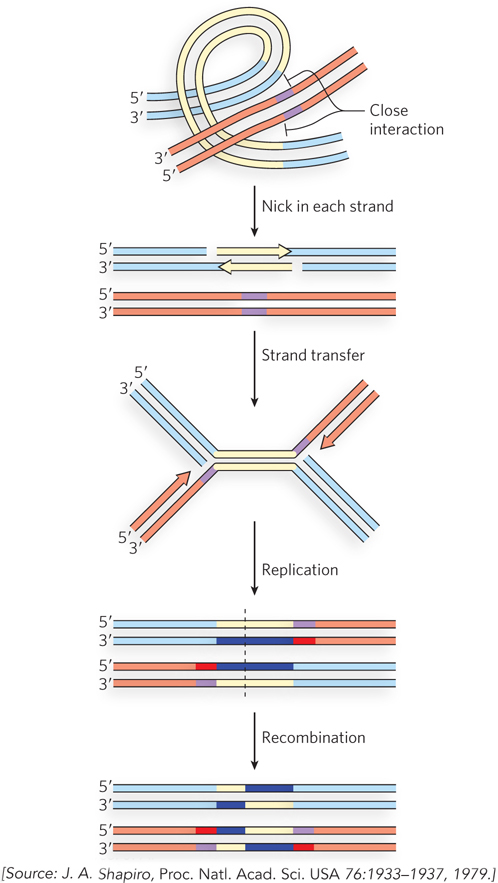
In 1979, James Shapiro, at the University of Chicago, proposed a mechanism for Mu transposition that laid out the main features of the process that we now know to occur (Figure 2). It involved nicking DNA strands to expose both 3′ ends of the transposon, then making a staggered break in the target DNA, leaving 5′ overhangs on the resulting ends. The transposon 3′ ends were then joined to the target 5′ ends. The remaining 3′ ends of the target would prime replication, creating two copies of the transposon and a cointegrate intermediate. This intermediate could be resolved by homologous or site-
Kiyoshi Mizuuchi, working with his associate Bob Craigie, found the intermediates. In the early 1980s, they developed an in vitro system that supported Mu transposition, using a plasmid that included a much-
515
There are two major steps in the process postulated by Shapiro: cleaving DNA and rejoining the ends to new partners, followed by replication (see Figure 2). We now know that the initial DNA cleavage and strand-
What had happened in the polyvinyl alcohol control? The absence of the polymer led to a partial reaction in which strand transfer became a dead end, with the resulting transposition intermediate building up to concentrations that made it much easier to detect and study. To confirm that this species was indeed a normal reaction intermediate (and not simply produced by unusual reaction conditions), Craigie and Mizuuchi isolated the putative intermediate DNA species, added back cell extract without MuA and MuB but with replication enzymes now aided by polyvinyl alcohol, and showed that the predicted transposition products were generated. The result was a definitive study establishing key facts about the pathway of Mu transposition. More broadly, the study played a major role in developing our current understanding of replicational transposition mechanisms.