Some Sets of Genes Are Regulated by Pre-mRNA Splicing in the Nucleus
Most eukaryotic genes, unlike bacterial genes, contain introns. Recent experiments conducted on Saccharomyces cerevisiae (budding yeast; referred to by nonscientists as baker’s yeast) suggest that pre-mRNA splicing is regulated in response to alterations in nutrient availability. Yeast cells grown in a nutrient-rich medium then shifted to a medium lacking essential amino acids rapidly reduce their level of splicing of transcripts for ribosomal proteins. This was detected by isolating total RNA from the cells at different time points during growth and starvation, then using DNA microarrays to determine changes in the mRNA population. Notably, only the r-protein gene transcripts were affected by the nutrient shift; the splicing of other transcripts was maintained, or even enhanced (Highlight 22-1).
Although the mechanism of such targeted splicing regulation has yet to be worked out, it seems that, like transcription, splicing provides eukaryotic cells with a means of rapidly adjusting expression levels of sets of genes in response to environmental triggers. This may be an important reason that eukaryotic cells have acquired and maintained introns over evolutionary time.
5′UTRs and 3′UTRs Coordinate the Translation of Multiple mRNAs
Sequence or structural elements in the untranslated regions of mRNAs provide a mechanism by which sets of transcripts can be controlled. One of the best-studied examples involves the regulation of iron concentration in mammalian cells. Although an essential element for cellular function, iron is also highly toxic, and its intracellular concentration must be carefully regulated.
A controlled level of iron relative to other cellular constituents, or iron homeostasis, is achieved through the coordinated expression of proteins responsible for iron uptake, storage, utilization, and export. Iron response proteins (IRPs) bind to a hairpin structure called the iron response element (IRE) in the 5′UTR or 3′UTR of the mRNAs encoding the proteins for iron uptake (the transferrin receptor), storage (ferritin), utilization (aconitase), and export (Fpn1). IRP-IRE complexes formed in the 5′UTR of an mRNA inhibit translation, probably by physically blocking access to ribosomes. In contrast, IRP-IRE complexes formed in the 3′UTR, as is the case for the transferrin receptor, prevent mRNA degradation (Figure 22-12). The IRE-binding activity of IRPs is high in iron-deficient cells and low in iron-rich cells. This switch in binding affinity results from iron-sulfur clusters that assemble in the IRPs and thereby block IRE binding only when iron is abundant. In this way, groups of genes in the iron-response pathway can be controlled together to bring about rapid changes in the relevant protein levels as cellular iron concentrations change.
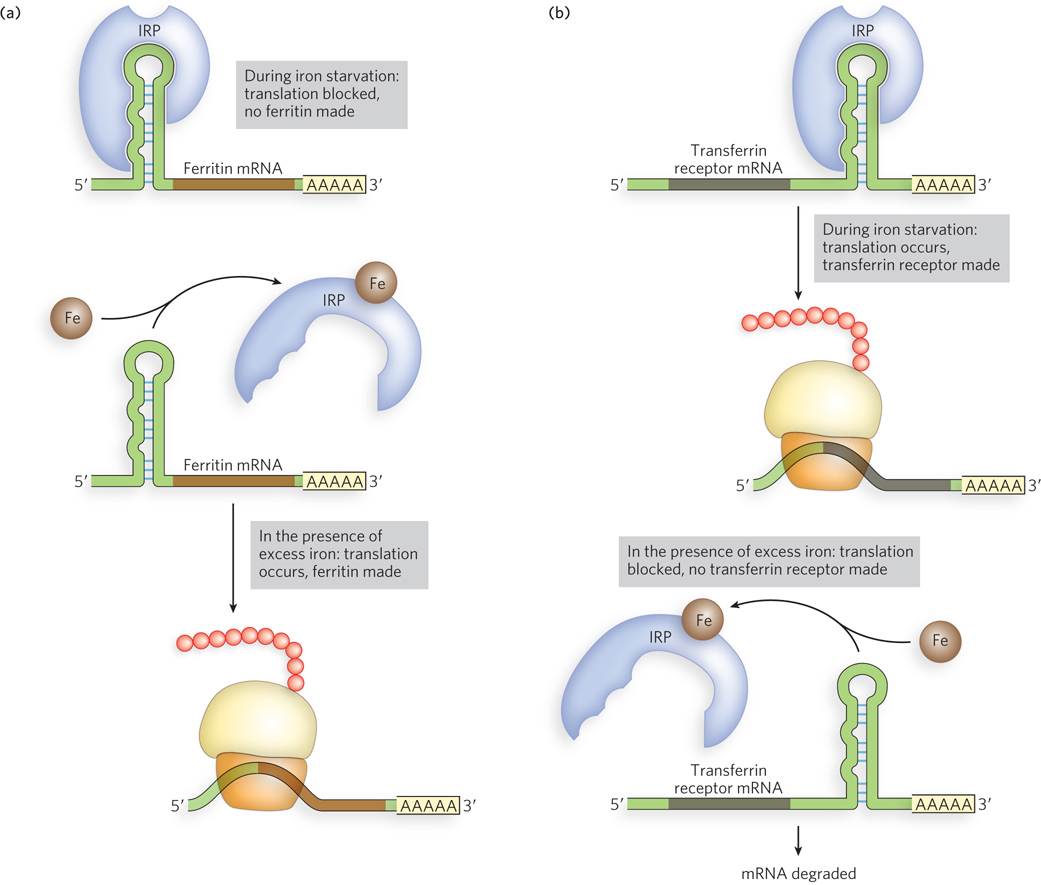
Figure 22-12: The function of iron response elements (IREs). In mammalian cells, IREs are bound by iron response proteins (IRPs) when iron levels are low, blocking the production of proteins that use and store iron (including ferritin), while promoting the production of proteins that facilitate iron uptake (the transferrin receptor). (a) IRP binding to the 5′UTR of ferritin mRNA inhibits translation (top). When iron is in excess (bottom), ferritin is produced. (b) IRP binding to the 3′UTR of the transferrin receptor mRNA stimulates translation (top); no transferrin receptor is made when iron is in excess (bottom).
HIGHLIGHT 22-1 EVOLUTION: Regulation of Splicing in Response to Stress

Christine Guthrie
The removal of introns from eukaryotic pre-mRNAs must occur before the mRNAs can be translated into protein. In higher eukaryotes, most genes have introns, and splicing regulates both when and where proteins are made. Furthermore, alternative splicing can produce different mRNAs and thus distinct proteins from a single initial transcript. In S. cerevisiae, however, only 5% of genes contain introns, and most of those genes have just one intron. All splice site sequences in yeast introns are very similar, and alternative splicing is rare. These observations led Christine Guthrie and her colleagues at the University of California, San Francisco, to wonder about the role of introns in the yeast genome: are introns maintained for a reason, perhaps to help cells respond to stressful situations?
To investigate this possibility, Guthrie and her students first examined the yeast genes that contain introns. They noticed that this set of genes includes many that code for metabolic regulators, such as enzymes that control the uptake and use of nutrients, and many ribosomal protein genes (RPGs). In fact, of the 139 RPGs in the yeast genome, 102 are interrupted by at least one intron. The RPGs are thus the largest functional category of intron-containing yeast genes.
Might these introns be playing a regulatory role in the expression of r-proteins? There were some hints that this might be the case. Previous research had shown that when yeast is starved for amino acids, which are essential for synthesizing new proteins, RPG transcription is suppressed and rRNA production and overall protein synthesis are reduced, whereas the production of enzymes required for amino acid biosynthesis is increased. Guthrie hypothesized that the splicing of RPG transcripts might be regulated in response to amino acid starvation.
To test this idea, Guthrie’s research team used DNA microarrays to examine the transcript-specific splicing changes resulting from exposure to two unrelated but environmentally relevant stresses: amino acid starvation and ethanol toxicity. RNA was purified from yeast cells at different times after exposure to each of these stresses and hybridized to microarrays containing DNA fragments representing the yeast genome. This analysis showed that splicing of the majority of RPGs is inhibited within minutes of induced amino acid starvation. By contrast, exposure to high levels of ethanol, which is not known to induce a global repression of translation, has little effect on RPG transcript splicing. Instead, in response to ethanol stress, the splicing of a different set of transcripts is reduced, and the splicing efficiency of a third group of transcripts is enhanced. The specificity of these responses and the speed of their onset—within minutes of stress induction—imply that splicing provides an important means of regulating gene expression in response to environmental stresses. This capacity for transcription-independent regulation could explain the evolutionary retention of introns in these yeast genes.
Since this initial study, several genome-wide surveys using microarray technology have shown distinct patterns of alternative splicing in various fruit fly and mammalian tissues. Furthermore, these experiments have identified RNA sequences that correspond to specific differential splicing events. These observations suggest that proteins controlling specific splicing events rely on an RNA code in their target transcripts and that this RNA code governs the correct set of differentially spliced mRNAs. Future research will focus on discovering the identities of these splicing regulators and understanding how they work together to bring about changes in pre-mRNA splicing in response to the cell’s needs.
Conserved AU-Rich Elements in 3′UTRs Control Global mRNA Stability for Some Genes
Large sets of genes can also be regulated by elements that affect the stability of RNA transcripts. Unlike bacterial mRNAs, many eukaryotic mRNAs are stable for hours or days. However, certain eukaryotic genes produce mRNAs with very rapid degradation rates. These genes are among those affected by a system for controlling mRNA stability that involves specific sequences in the 3′UTRs.
In mammalian cells, mRNAs with sequence elements rich in A and U nucleotides, called AU-rich elements (AREs), are targeted for rapid degradation. The most common ARE motif is AUUUA, and it is often part of a larger AU-rich region such as WWWUAUUUAUUUW, where W is A or U.
ARE-containing mRNAs typically encode proteins that regulate either cell growth or an organism’s response to infection, inflammation, and environmental stimuli. For example, mRNAs transcribed from proto-oncogenes (many of which encode proteins that promote cell division) have a very short half-life in the cell. In resting or unstimulated cells, ARE-dependent degradation ensures there is only low-level expression of these potent proteins. When cells respond to particular signals, ARE-containing mRNAs are bound by certain RNA-binding proteins, especially those in the ELAV (embryonic lethal abnormal visual) family, first discovered in Drosophila. Thus bound, the mRNAs become more stable, and the expression levels of the proteins they encode increase. Notably, in cancerous or chronically inflamed human cells, this ARE-dependent regulation goes awry. This can be caused by increased levels of ELAV family proteins, such as HuR (“Hu” refers to an antibody, sometimes expressed in tumor-associated neurological disorders, that binds ELAV proteins). The increased mRNA stability conferred by the binding of HuR and related proteins allows increased expression of proteins that normally should not be present in cells, contributing to abnormal cell physiology and/or suppression of pathways that control cell growth.
One of the first demonstrations that an ARE sequence controls mRNA stability came from a study of genes for cytokines, cell proliferation factors produced principally by immune system cells that have very unstable mRNAs. In a comparison of the human and mouse gene sequences for a lymphokine (a type of cytokine) known as granulocyte-monocyte colony stimulating factor (GM-CSF), the most conserved sequence was outside the protein-coding region, in the 3′UTR. The protein-coding sequence was only 65% conserved, but a 51 nucleotide AT-rich sequence in the 3′UTR was 93% conserved, suggesting that the 3′UTR sequence plays a functional role. The sequence is also conserved in many other cytokine genes and proto-oncogenes. To test these 3′UTR sequence for function in mRNA stability, the AT-rich sequence in the 3′UTR of the GM-CSF gene was cloned into the 3′UTR of the human β-globin gene. The stability of the resultant mRNA was compared with that of the wild-type globin gene and that of a globin gene into which a GC-rich sequence (from the GM-CSF gene) was inserted. The AT-rich element in the 3′UTR caused a marked loss of mRNA stability (Figure 22-13).
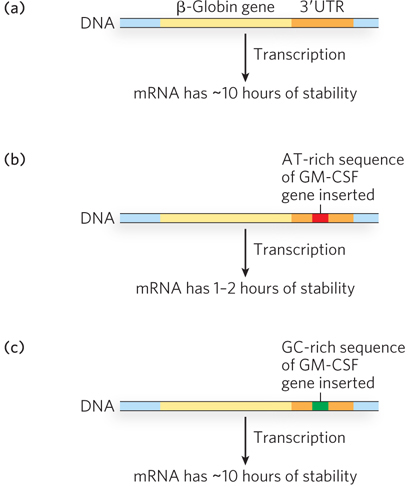
Figure 22-13: AU-rich elements (AREs) affect mRNA stability. AREs are binding sites for certain proteins that either stabilize or destabilize the bound mRNA. (a) mRNA transcribed from the β-globin gene is stable for about 10 hours. (b) When a single AT-rich ARE sequence is introduced into the globin gene, such as that derived from the GM-CSF gene, mRNA stability is greatly reduced. (c) When a GC-rich sequence is introduced into the gene, mRNA stability is restored.
With the human genome sequenced, we now know that 5% to 8% of human genes encode ARE-regulated mRNAs. Numerous ARE-binding proteins have been identified, in addition to the ELAV family proteins. As in the IRP-IRE system described above, these ARE-binding proteins can bind multiple mRNAs. In contrast to ELAV family proteins, which seem to increase mRNA stability, some ARE-binding proteins recruit bound mRNAs to the cell’s 3′-deadenylation and 5′-decapping machinery, leading to rapid degradation of the transcript. When cells require increased amounts of the proteins encoded by ARE-containing mRNAs, the levels of destabilizing ARE-binding proteins decline and binding efficiencies drop. A concomitant increase in ELAV family proteins may occur, thereby stabilizing ARE-containing mRNAs and ensuring more translation from those transcripts.
SECTION 22.3 SUMMARY
Cells often regulate large sets of genes together to bring about changes in cell fate.
Pre-mRNA splicing provides one mechanism by which cells control the expression of sets of genes. In yeast, splicing of many r-protein RNA transcripts is regulated in response to changes in nutrient availability.
Both mRNA stability and translational efficiency are controlled by elements in the untranslated regions of some mRNAs.
In mammalian cells, the expression of proteins involved in iron homeostasis is controlled by the iron response element (IRE), a structure in the 5′UTR or 3′UTR of specific mRNAs. In the presence of iron, IREs bind iron response proteins (IRPs), triggering increased or decreased expression, depending on the location of the IRE in the mRNA transcript.
About 5% to 8% of mammalian mRNAs contain AU-rich elements (AREs) in their untranslated regions that bind ARE-binding proteins, leading to either stabilization (if ELAV family proteins) or rapid degradation (if destabilizing ARE-binding proteins) of the mRNA. When cells require a change in ARE-regulated protein levels in response to infection or other stimuli, ARE-binding activity is altered and expression levels increase or decrease.