22.6 FINALE: MOLECULAR BIOLOGY, DEVELOPMENTAL BIOLOGY, AND EVOLUTION
Molecular biology is a story of biological information—
Each topic in molecular biology has evolutionary significance. Errors or random events in DNA replication, recombination, and repair fuel genomic changes—
The Interface of Evolutionary and Developmental Biology Defines a New Field
South America has several species of seed-
The diversity of living creatures on our planet was a source of wonder for humans long before scientists sought to understand its origins. The extraordinary insight handed down to us by Darwin, inspired in part by his encounter with the Galápagos finches, provided a broad explanation for the existence of organisms with a vast array of appearances and characteristics. It also gave rise to many questions about the mechanisms underlying evolution. Answers to those questions have started to appear, first through the study of genomes and nucleic acid metabolism in the last half of the twentieth century, and more recently through an emerging field nicknamed evo-
In its modern synthesis, the theory of evolution has two main elements: mutations in a population generate genetic diversity, and natural selection then acts on this diversity to favor individuals with more useful genomic tools and to disfavor others. Mutations occur at significant rates in every individual’s genome, in every cell (see Chapters 3 and 12). Advantageous mutations in single-
792
Modern genomics has revealed that the human genome contains fewer genes than expected—
Small Genetic Differences Can Produce Dramatic Phenotypic Changes
The kinds of mutant organisms shown in Figure 22-30 were studied by the English biologist William Bateson in the late nineteenth century. Bateson used his observations to challenge the Darwinian notion that evolutionary change would have to be gradual. Recent studies of the genes that control organismal development have strongly supported Bateson’s ideas. Subtle changes in regulatory patterns during development, reflecting just one or a few mutations, can result in startling physical changes and fuel surprisingly rapid evolution.
The Galápagos finches provide a wonderful example of the link between evolution and development. There are at least 14 species (some specialists list 15), and they are distinguished in large measure by their beak structure. The ground finches, for example, have broad, heavy beaks adapted to crushing hard, large seeds. The cactus finches have longer, slender beaks, ideal for probing cactus flowers and fruit (Figure 22-33).
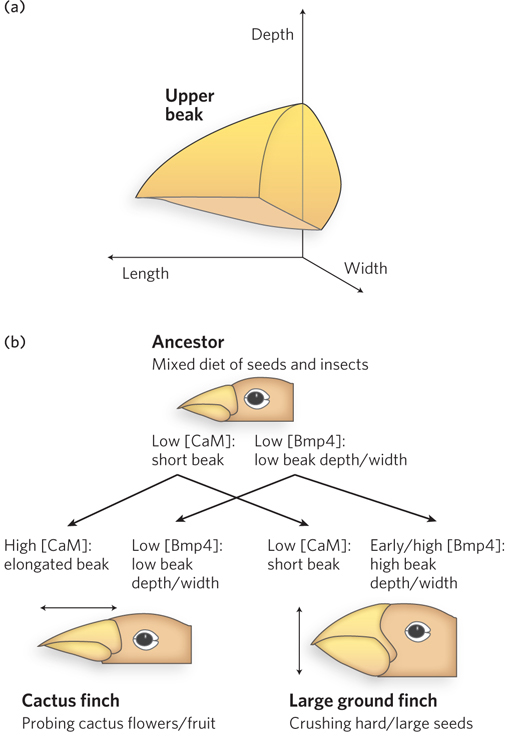
Clifford Tabin and his colleagues carefully surveyed a set of genes expressed during avian craniofacial development. They identified a single gene, Bmp4, whose expression level correlated with the formation of the more robust beaks of the ground finches. More robust beaks were also formed in chicken embryos when high levels of the Bmp4 protein were artificially expressed in the appropriate tissues, confirming the importance of Bmp4. In a similar study, the formation of long, slender beaks was linked to the expression of the protein calmodulin in particular tissues at appropriate developmental stages. Thus, major changes in the shape and function of the beak can be brought about by subtle changes in the expression of just two genes involved in developmental regulation. Very few mutations are required, and the needed mutations affect regulation. New genes are not required. Note that Bmp4 is a member of a family of signaling proteins, with roles in development similar to those of the Wnt and Hedgehog proteins. Like Wnt and Hedgehog, Bmp homologs are widely conserved in eukaryotes. And as in the other signaling pathways, alterations in Bmp signaling pathways can have large effects on development.
The system of regulatory genes that guides development is remarkably conserved among all vertebrates. Elevated expression of Bmp4 in the right tissue at the right time leads to more robust jaw parts in zebra fish. The same gene plays a key role in tooth development in mammals. The development of eyes is triggered by the expression of a single gene, Pax6, in fruit flies and in mammals. The mouse Pax6 gene will trigger the development of fruit fly eyes in the fruit fly, and the fruit fly Pax6 gene will trigger the development of mouse eyes in the mouse. In each organism, these genes are part of the much larger regulatory cascade that ultimately creates the correct structures in the correct locations in each organism. The cascade is ancient; the Hox genes have been part of the developmental program of multicellular eukaryotes for more than 500 million years. Subtle changes in the cascade can have large effects on development, and thus on the ultimate appearance of the organism. These same subtle changes can promote rapid evolution. For example, the 400 to 500 described species of cichlids (spiny-
793
Our discussion of developmental regulation brings us full circle, back to a biochemical beginning—
SECTION 22.6 SUMMARY
Developmental biology and evolutionary biology are closely related. The two fields inform each other, and molecular biology is intimately intertwined with both.
Major changes in the appearance and/or function of multicellular eukaryotes can be effected by subtle changes in an organism’s developmental program, involving mutations in the regulatory genes that guide the process.
UNANSWERED QUESTIONS
Our understanding of gene regulation remains incomplete in many areas. Indeed, the recent discovery of RNA interference, which has proved to be a major mode of regulation, underscores the likelihood that more fundamentals remain to be elucidated.
How is alternative splicing regulated? Mounting evidence suggests that alternative splicing accounts for a much greater degree of protein complexity in higher eukaryotes than would be predicted by simply counting the number of open reading frames in a genome. How such alternative splicing is regulated is not well understood, nor have mechanisms of tissue-
specific splicing been worked out. How and when do miRNAs control gene expression in human cells? The human genome encodes several hundred miRNAs, yet the targets and functions of most of these are currently unknown. How do we harness these newly discovered regulatory mechanisms to provide new therapies for cancer and other diseases?
What other regulatory mechanisms have we just not uncovered yet? RNA interference is a fairly recent discovery. What are the functions of all the new RNAs being discovered in eukaryotic genomes?
Are transcriptional and posttranscriptional steps in gene expression coordinately regulated? What kinds of mechanisms might enable communication between the cytoplasm and the nucleus to adjust transcription, splicing, and mRNA transport rates in response to increased or decreased translation of a particular mRNA?
What are all of the signals that guide the action of regulatory proteins and the development of specific tissues? A much more detailed understanding is needed to completely unleash the potential of stem cell technologies, particularly with respect to the control of differentiation of induced pluripotent stem cells. That potential includes new cancer treatments, the regeneration of lost limbs, and the replacement of diseased tissues (e.g., heart, lung, kidney) without the danger of tissue rejection.
794
A Natural Collaboration Reveals a Binding Protein for a 3′UTR
Zhang, B., M. Gallegos, A. Puoti, E. Durkin, S. Fields, J. Kimble, and M.P. Wickens. 1997. A conserved RNA-

Scientific collaborations come about in many ways, as the discovery of one gene-
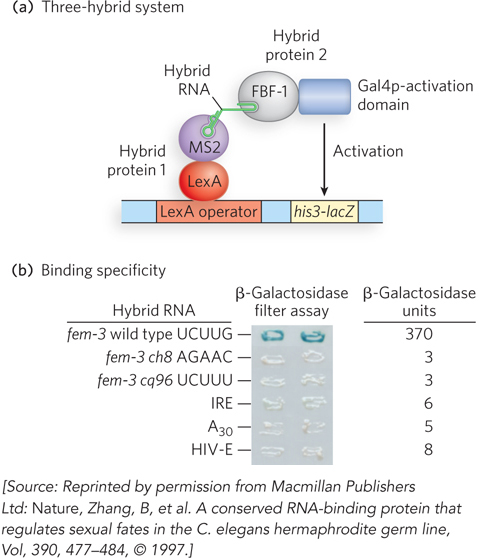
In 1996, Marvin Wickens and his coworkers reported the invention of the three-
The investigators initiated a screen of a cDNA library in which C. elegans genes were fused to the gene encoding the Gal4p activation domain. The new three-
RNAi experiments confirmed the role of FBF proteins in germ cell fate. Studies of the FBF proteins quickly became part of a still expanding effort to characterize the function of PUF family proteins.
795
Little RNAs Play a Big Role in Controlling Gene Expression
Lagos-
Lau, N.C., L.P. Lim, E.G. Weinstein, and D.P. Bartel. 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294:858–
Lee, R.C., and V. Ambros. 2001. An extensive class of small RNAs in Caenorhabditis elegans. Science 294:862–
Scientific discovery has a way of occurring in bursts of insight, often with input from multiple research teams whose ideas and experiments converge on a new line of thinking. In the field of RNA interference, such a conceptual breakthrough occurred in 2001 with the finding by three different labs that small regulatory RNA molecules are abundant in eukaryotic cells. Scientists had come to suspect that small RNAs might normally be produced in cells as a means of controlling gene expression. This suspicion was based on the discovery by Craig Mello and Andrew Fire that double-
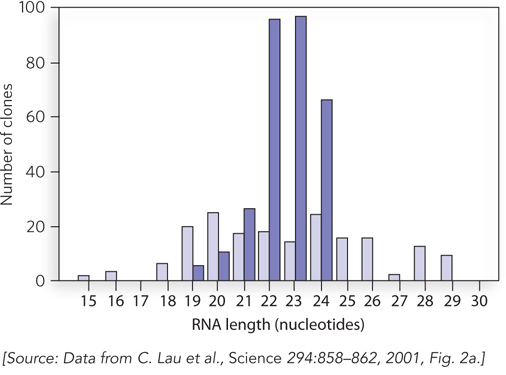
Each team took a similar experimental approach, in which C. elegans or mammalian cells were grown in the laboratory and total cellular RNA was isolated. The total RNA was fractionated by size to enable purification of RNA molecules about 20 to 30 nucleotides long, the size of the molecules used in the Mello and Fire experiments. To identify these molecules, the RNAs were covalently linked at their 3′ ends to oligonucleotide sequences that provided binding sites for a complementary oligonucleotide, which could be used to prime the reverse transcription of the RNA into DNA. The complementary strand of this DNA sequence could be produced in a similar fashion, by covalently attaching it to a second oligonucleotide of defined sequence at its 3′ end. Once the small RNAs had been copied into double-
The sequences of these small RNAs proved very exciting, because in many cases they were complementary to sequences found in the host genome. This finding suggested that the small RNAs were produced as part of a large regulatory pathway in which small RNA molecules, dubbed microRNAs (miRNAs), could base-
Why were miRNAs overlooked by molecular biologists for so long? One reason is simply size: because they are so small, they tended to be ignored or were thought to be irrelevant degradation products rather than functional RNAs produced by the cells.
796
Everything Old Is New Again: Beauty at the Turn of a Developmental Switch
Carroll, S.B., J. Gates, D.N. Keys, S.W. Paddock, G.E. Panganiban, J.E. Selegue, and J.A. Williams.1994. Pattern formation and eyespot determination in butterfly wings. Science 265:109–

We might not notice the diminutive fruit fly, but butterflies rarely fail to inspire fascination. The bold colors and patterns in a butterfly wing that catch our eye—
Sean Carroll’s boyhood fascination with butterflies was eventually translated into research in a lab at the University of Wisconsin, where he studies insect development. In the early 1990s, it was already clear that many genes that control development are highly conserved, not just in insects, but in all higher eukaryotes. In setting out to decipher the development of butterfly wing patterns, Carroll decided that the genes known to affect the development of Drosophila wings were a good place to start.

Carroll’s subject was the butterfly Precis coenia, also called the Buckeye, found each summer over much of the United States. His laboratory succeeded in cloning a series of P. coenia genes homologous to Drosophila genes known to control wing development, including the genes for signaling proteins called Wingless and Decapentaplegic, and transcription factors called Apterous, Invected, Scalloped, and Distal-
One day, Carroll’s student, Julie Gates, called him over because she saw a pattern of genes turned on in so-

797