Concept 12.1: There Are Powerful Methods for Sequencing Genomes and Analyzing Gene Products
Genome sequencing involves determining the nucleotide base sequence of the entire genome of an organism. For a prokaryote with a single chromosome, the genome sequence is one continuous string of base pairs (bp). In a eukaryote, there are separate sequences for each chromosome. Scientists can use this genomic information in several ways:
- The genomes of different species can be compared to find out how they differ at the DNA level, and this can be used to trace evolutionary relationships.
- The sequences of individuals within a species and even tissues within an organism can be compared to identify mutations that affect particular phenotypes.
- The sequence information can be used to identify particular traits, such as genes associated with diseases.
The notion of sequencing the entire genome of a complex organism was not contemplated until 1986. The Nobel laureate Renato Dulbecco and others proposed at that time that the world scientific community be mobilized to undertake the sequencing of the entire human genome. One motive was to detect DNA damage in people who had survived the atomic bomb attacks and been exposed to radiation in Japan during the Second World War. But in order to detect changes in the human genome, scientists first needed to know its normal sequence.
The result was the publicly funded Human Genome Project, an enormous undertaking that was successfully completed in 2003. This effort was aided and complemented by privately funded groups. The project benefited from the development of many new methods that were first used in the sequencing of smaller genomes—those of prokaryotes and simple eukaryotes, the model organisms you are familiar with from studies in genetics and cell biology. Many of these methods are still applied widely, and powerful new methods for sequencing genomes have emerged. These are complemented by new ways to examine phenotypic diversity in a cell’s proteins and in the metabolic products of the cell’s enzymes.

Go to ANIMATED TUTORIAL 12.1 Sequencing the Genome
PoL2e.com/at12.1
Methods have been developed to rapidly sequence DNA
Many prokaryotes have a single chromosome, whereas eukaryotes have many. Because of their differing sizes, chromosomes can be separated from one another, identified, and experimentally manipulated. It might seem that the most straightforward way to sequence a chromosome would be to start at one end and simply sequence the DNA molecule one nucleotide at a time. The task is somewhat simplified because only one of the two strands needs to be sequenced, the other being complementary. However, this large-polymer approach is not practical. Using current methods, only several hundred bp can be sequenced at a time, whereas a human chromosome (for example) may be hundreds of millions of bp long.
As you will see, the key to determining genome sequences is to perform many sequencing reactions simultaneously, after first breaking the DNA up into millions of small, overlapping fragments.
In the 1970s, Frederick Sanger and his colleagues invented a way to sequence DNA by using chemically modified nucleotides that were originally developed to stop cell division in cancer. Variations of this method were used to obtain the first human genome sequence as well as those of several model organisms. However, it was relatively slow, expensive, and labor-intensive. The first decade of the new millennium saw the development of faster and less expensive methods, often referred to under the general term high-throughput sequencing. These methods use miniaturization techniques first developed for the electronics industry, as well as the principles of DNA replication and the polymerase chain reaction (PCR).
LINK
You can review the processes of DNA replication and PCR in Concept 9.2
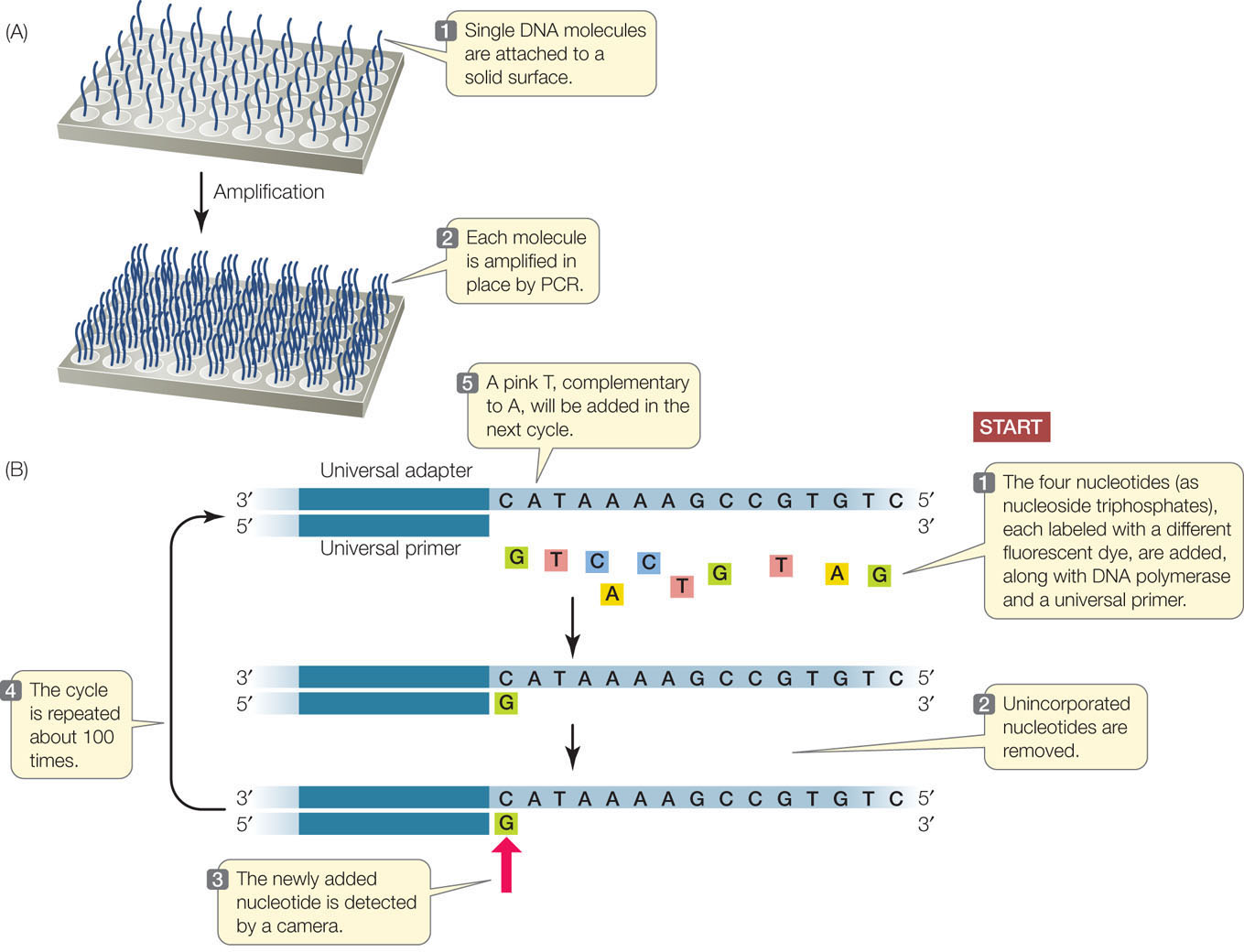
High-throughput sequencing methods are rapidly evolving. Just one of the many approaches is outlined here and illustrated in FIGURE 12.1. First the DNA is prepared for sequencing:

Go to ANIMATED TUTORIAL 12.2 High-Throughput Sequencing
PoL2e.com/at12.2
- A large molecule of DNA is cut into small fragments of about 100 bp each. This can be done physically, using mechanical forces to shear (break up) the DNA, or by using enzymes that hydrolyze the phosphodiester bonds between nucleotides at intervals in the DNA backbone.
- The DNA is denatured by heat, breaking the hydrogen bonds that hold the two strands together. Each single strand acts as a template for the synthesis of new, complementary DNA.
- Short, synthetic adapter sequences (oligonucleotides) are attached to each end of each fragment, and the fragments are attached to a solid support. The support can be a microbead or a flat surface.
- Primers complementary to the adapters are used in PCR reactions to produce many (approximately 1,000) copies of each DNA fragment. The multiple copies at a single location allow for easy detection of added nucleotides during the sequencing steps.
Once the DNA has been attached to a solid substrate and amplified, it is ready to be used as a template for sequencing (see Figure 12.1B):
- At the beginning of each sequencing cycle, the DNA fragments are heated to denature them. A universal primer, DNA polymerase, and the four deoxyribonucleoside triphosphates (dNTPs: dATP, dGTP, dCTP, and dTTP) are added. Each of the four nucleotides (i.e., the dNTPs) is tagged with a different fluorescent dye. The universal primer is complementary to the adapter sequence at one end of each DNA fragment.
- The DNA sequencing reaction is set up so that only one nucleotide at a time is added to the new DNA strand, which is complementary to the template strand. After each addition, the unincorporated nucleotides are removed.
- The fluorescence of the new nucleotide at each location is detected with a camera. The color of the fluorescence indicates which of the four nucleotides was added.
- The fluorescent tag is removed from the nucleotide that is already attached, and then the sequencing cycle is repeated. Images are captured after each nucleotide is added. The series of colors at each location indicates the sequence of nucleotides in the growing DNA strand at that location.
The power of this method derives from several factors:
- It is fully automated and miniaturized.
- Millions of different fragments are sequenced at the same time.
- It is an inexpensive way to sequence large genomes. For example, at the time of this writing, a complete human genome could be sequenced in a few days for several thousand dollars. In contrast, the Human Genome Project took 13 years and $2.7 billion to sequence one genome!
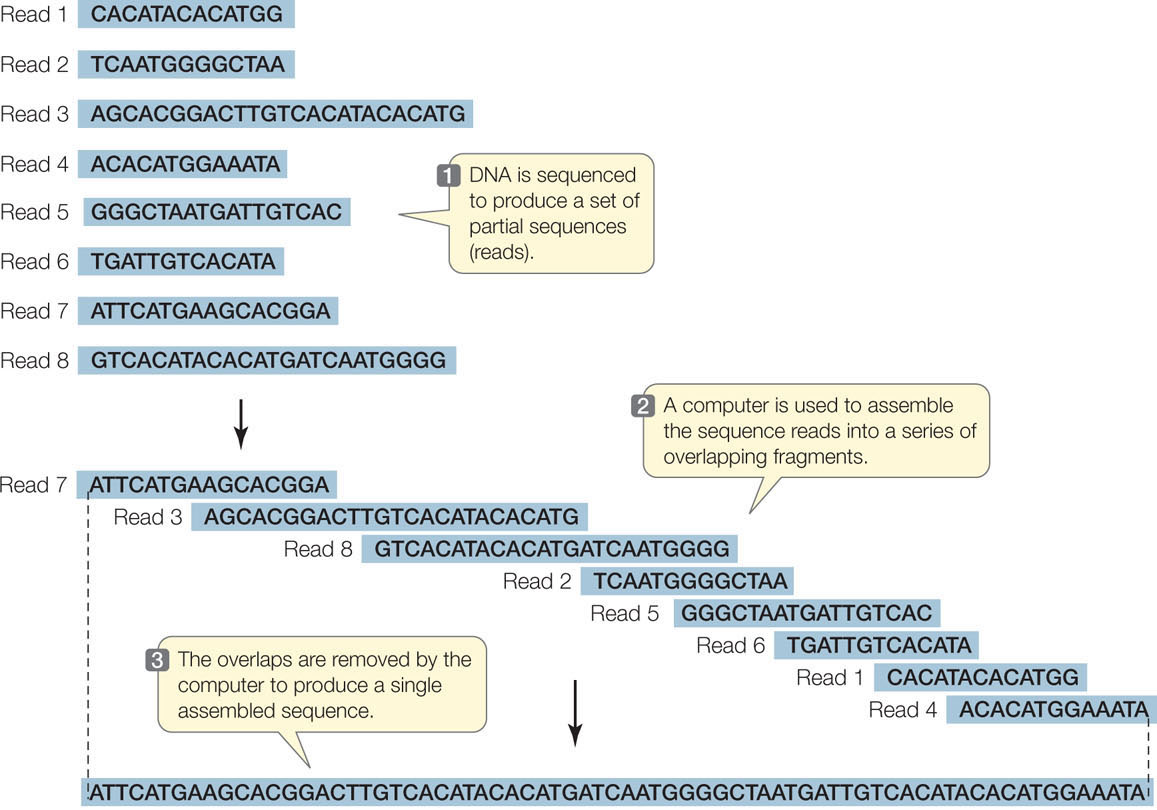
The technology used to sequence millions of short DNA fragments is only half the story, however. Once these sequences have been determined, the problem becomes how to put them together. In other words, how are they arranged in the chromosomes from which they came? Imagine if you cut out every word in this book (there are about 500,000 of them), put them on a table, and tried to arrange them in their original order! The enormous task of determining DNA sequences is possible because the original DNA fragments are overlapping.
Let’s illustrate the process using a single 10-bp DNA molecule. (This is a double-stranded molecule, but for convenience we show only the sequence of the noncoding strand.) The molecule is cut three ways using three different enzymes. The first cut generates the fragments:
TG, ATG, and CCTAC
The second cut of the same molecule generates the fragments:
AT, GCC, and TACTG
The third cut results in:
CTG, CTA, and ATGC
237
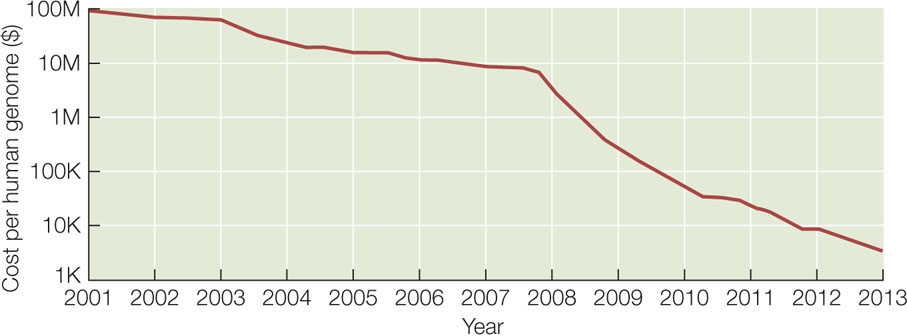
Can you put the fragments in the correct order? (The answer is ATGCCTACTG.) For genome sequencing, the fragments are called “reads” (FIGURE 12.2). Of course, the problem of ordering 2.5 million fragments of 100 bp from human chromosome 1 (246 million bp) is more challenging than our 10-bp example above. The field of bioinformatics was developed to analyze DNA sequences using complex mathematics and computer programs. The cost of genome sequencing and analysis has gone down rapidly:
Genome sequences yield several kinds of information
New genome sequences are being published at an accelerating pace, creating a torrent of biological information. This information is used in two related fields of research, both focused on studying genomes. In functional genomics, biologists use sequence information to identify the functions of various parts of genomes (FIGURE 12.3). These parts include:
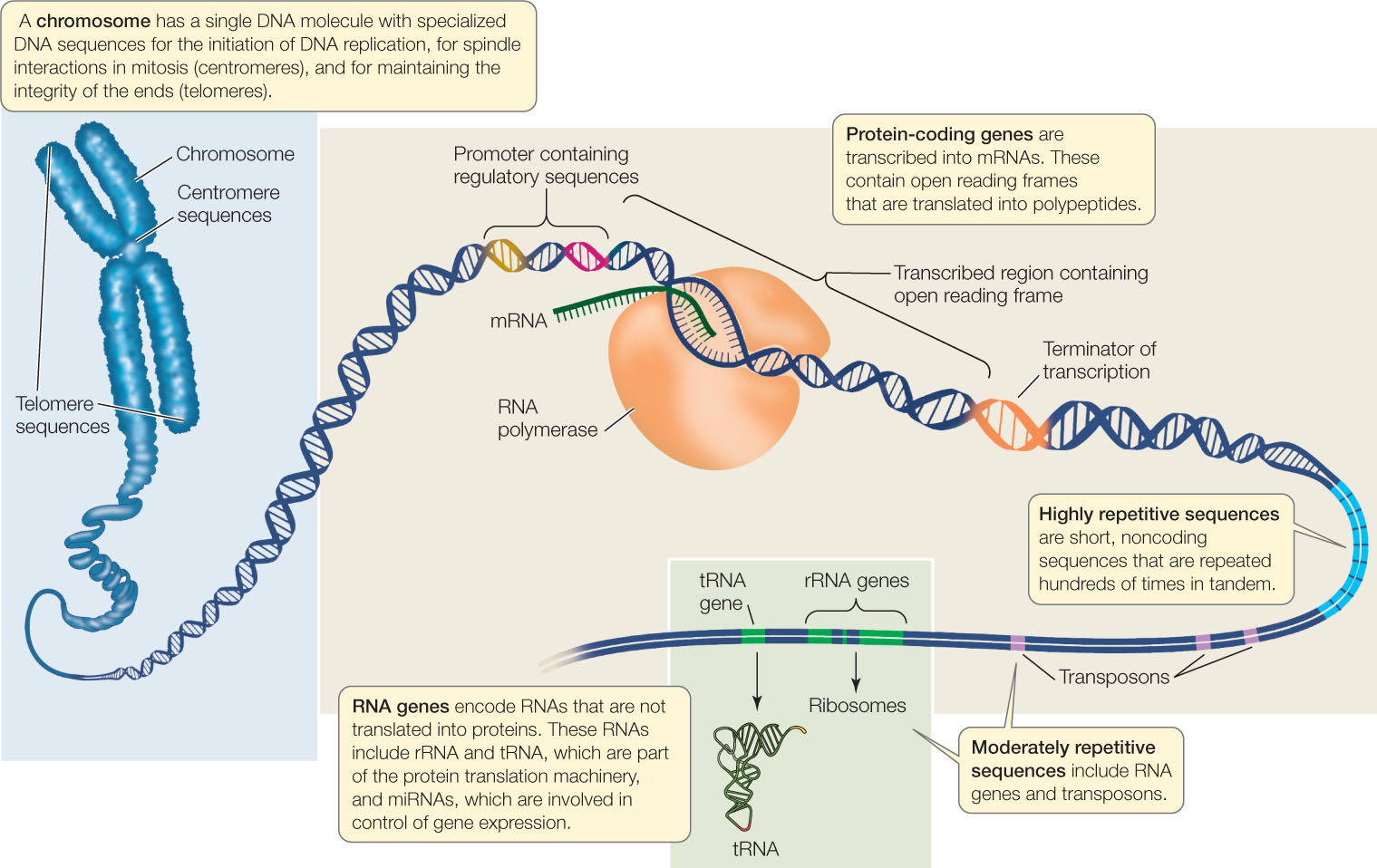
- Open reading frames, which are sequences of DNA that contain no stop codons, and thus may encode parts of proteins. An open reading frame that begins with a start codon or an intron consensus sequence (boundary between exon and intron) and ends with a stop codon or an intron consensus sequence may be an exon, which encodes part or all of a polypeptide. See Concepts 10.2 and 10.3 to review introns, exons, and the genetic code.
- Regulatory sequences, such as promoters and terminators for transcription. These are identified by their proximity to open reading frames and because they contain consensus sequences for the binding of specific transcription factors and RNA polymerase.
- Regions of DNA that have regulatory sequences at each end and one or more open reading frames; these may be protein-coding genes. The amino acid sequence of a protein can be deduced by applying the genetic code (see Figure 10.11) to the DNA sequences of the open reading frames within the gene. A major goal of functional genomics is to identify, and understand the function of, every protein-coding gene in each genome.
- RNA genes, including genes for rRNA, tRNA, and miRNA (see Concept 11.4).
- Other noncoding sequences that can be classified into various categories, including centromeric regions (see Concept 7.2), telomeric regions (see Concept 9.2), transposons (see Concept 12.2), and other repetitive sequences.
238
Functional regions in a newly described genomic sequence can be identified by searching DNA databases for similar or identical sequences in other organisms. There are now massive databases (accessible online) containing DNA sequences and their known or possible functions.
Sequence information is also used in comparative genomics: the comparison of a newly sequenced genome (or parts thereof) with sequences from other organisms. This can provide further information about the functions of sequences and can be used to trace evolutionary relationships among different organisms.
LINK
The application of genome sequencing to reconstructing phylogenies (evolutionary trees) is described in Concept 16.2
Phenotypes can be analyzed using proteomics and metabolomics
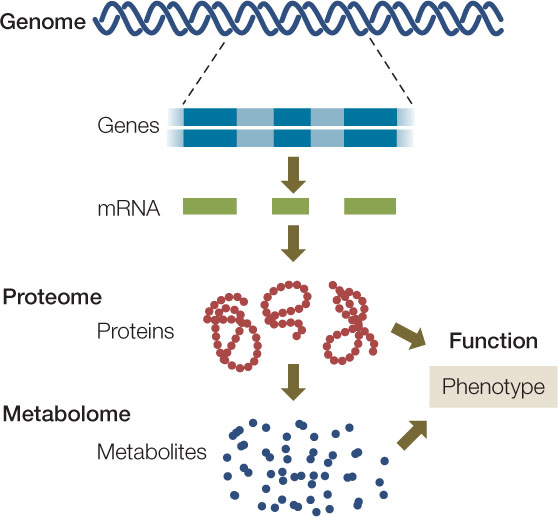
“The human genome is the book of life.” Statements like this were common when the human genome sequence was first revealed. They reflect the concept of genetic determinism—the idea that a person’s phenotype is determined solely by his or her genotype. But is an organism just a product of gene expression? We know that it is not. The proteins and small molecules present in any cell at a given point in time reflect not just gene expression but modifications caused by the intracellular and extracellular environments. Two new fields have emerged to complement genomics and take a more complete snapshot of a cell or organism: proteomics and metabolomics.
PROTEOMICS
Many genes encode more than a single protein (FIGURE 12.4A). As we described in Concept 11.4, alternative splicing leads to different combinations of exons in the mature mRNAs transcribed from a single gene. Posttranslational modifications also increase the number and the structural and functional diversity of proteins derived from one gene (see Figures 12.4A and 10.21). The proteome is the sum total of the proteins produced by an organism, and it is more complex than the organism’s genome.
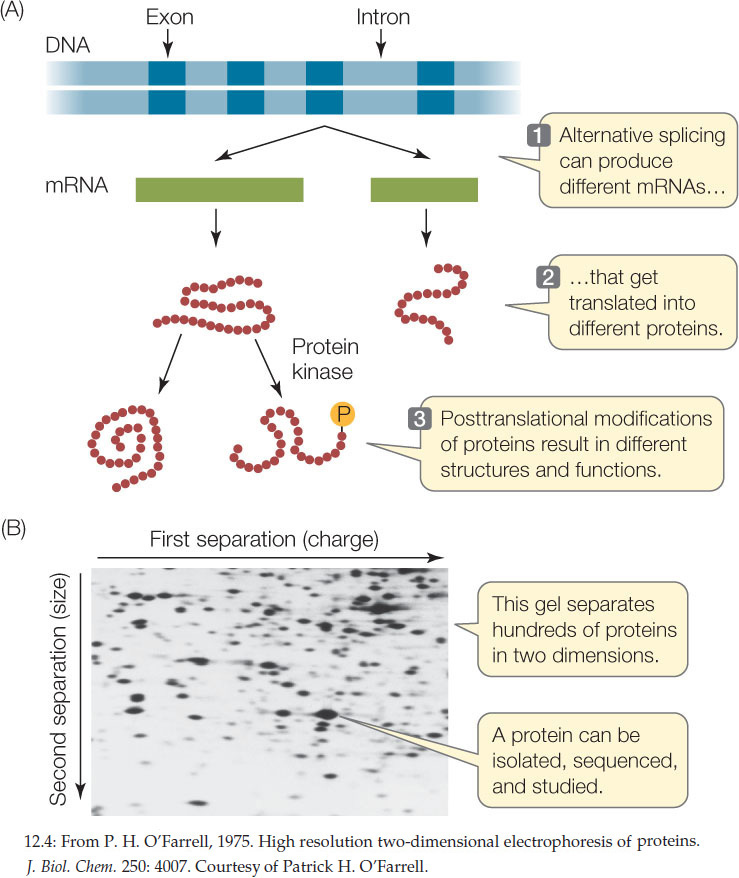
Several approaches are commonly used to analyze proteins and the proteome:
- Because of their unique amino acid compositions (primary structures), most proteins have unique combinations of electric charge and size. On the basis of these two properties, they can be separated by two-dimensional gel electrophoresis (FIGURE 12.4B).
- Once they have been isolated, individual proteins can be analyzed by mass spectrometry. This technique uses electromagnets to identify molecules by the masses of their atoms, and it can also be used to determine the structures of molecules.
- Antibodies can also be used to isolate specific proteins, or to detect the proteins in cells or tissues.
239
Whereas genomics seeks to describe the genome and its expression, proteomics seeks to identify and characterize all of the expressed proteins. Its ultimate aim is just as ambitious as that of genomics. Comparisons of proteomes among organisms have revealed common sets of proteins that can be categorized into groups with similar amino acid sequences and functions. Often these share three-dimensional structural regions called domains (for example, the heme-binding domain of hemoglobin). While a particular organism may have many unique proteins, those proteins are often just unique combinations of domains that exist in proteins of other organisms. This reshuffling of the genetic deck is a key to evolution.
Metabolomics
Studying genes and proteins gives a limited picture of what is going on in a cell. But as we have seen, both gene function and protein function are affected by a cell’s internal and external environments. Many proteins are enzymes, and their activities affect the concentrations of their substrates and products. So as the proteome changes, so do the abundances of these (often small) molecules, called metabolites. The metabolome is the complete set of small molecules present in a cell, tissue, or organism. These include:
- Primary metabolites that are involved in normal processes, such as intermediates in pathways such as glycolysis.
This category also includes hormones and other signaling molecules. - Secondary metabolites, which are often unique to particular organisms or groups of organisms. They are often involved in special responses to the environment. Examples are antibiotics made by microbes, and the many chemicals made by plants that are used in defense against pathogens and herbivores.
Metabolomics aims to describe the metabolic profile of a tissue or organism under particular environmental conditions. Measuring metabolites involves sophisticated analytical instruments. If you have studied organic or analytical chemistry, you may be familiar with gas chromatography and high-performance liquid chromatography, which are used to separate molecules with different chemical properties. Mass spectrometry and nuclear magnetic resonance spectroscopy are used to identify molecules. These measurements result in “chemical snapshots” of cells or organisms, which can be related to physiological states.
Plant biologists are far ahead of medical researchers in the field of metabolomics. Tens of thousands of secondary metabolites, many of them made in response to environmental challenges, have been identified in plants. The metabolomes of agriculturally important plants are being described, and this information may be important in optimizing plant growth for food production.
LINK
Some of the many secondary metabolites made by plants are discussed in Concept 28.2
Taken together, the genome, proteome, and metabolome can move biologists toward a more comprehensive picture of an organism’s genotype and phenotype (FIGURE 12.5).
240
CHECKpoint CONCEPT 12.1
- Using a table, compare genomics, proteomics, and metabolomics with regard to the methods used and results obtained.
- A DNA molecule is cut into the following fragments that are sequenced: AGTTT, TAGG, CGAT, and CCT. The same molecule is cut in a different way to produce TTCGA, TCCT, AGT, GG, and TA. A third cut produces TTCGAT, CCTT, AGG, and AGT. What is the sequence of this DNA?
- If you were designing a computer program to recognize important sequences in DNA, what types of sequences would you include? What would you learn from finding these sequences in the DNA? Be as specific as you can.
The first cellular genomes to be fully sequenced were those of prokaryotes. In the next concept we will discuss these relatively small, compact genomes.