Concept 12.4: The Human Genome Sequence Has Many Applications
During the first decade of this millennium the haploid genomes of more than ten individuals were sequenced and published. With the rapid development of new sequencing technologies, the time is approaching when a human genome can be sequenced for less than $1,000.
The human genome sequence held some surprises
The following are just some of the interesting facts we have learned about the human genome:
- Among the 3.2 billion bp in the haploid human genome, there are about 21,000 protein-coding genes. This was a surprise. Before sequencing began, the diversity of human proteins suggested there would be 80,000–150,000 genes. The actual number—not many more than in a nematode—means that posttranscriptional mechanisms (such as alternative splicing) must account for the observed number of proteins in humans. It turns out that most human genes encode multiple proteins.
- The average protein-coding gene spans 27,000 bp, and virtually all genes have many introns. Gene sizes vary greatly, from about 1,000 to 2.4 million bp. Variation in gene size is to be expected given that human proteins vary in size, from about 100 to 5,000 amino acids per polypeptide chain.
- More than 50 percent of the genome is made up of transposons and other repetitive sequences. Most transposons are inactive most of the time.
- About 75 percent of the genome is transcribed at some point in some cells. This result came from a recent analysis of genome expression in human cells in culture. A typical specialized cell only transcribes about 25 percent of its genome, so most transcripts are cell-type specific. Many transcripts are noncoding RNAs involved in regulating gene expression.
- Most of the genome (at least 99 percent) is the same in all people. Despite this apparent homogeneity, there are, of course, many individual differences. Current estimates suggest that each haploid genome has variations in about 3.3 million single nucleotide polymorphisms (SNPs; see below), as well as short repeated sequences that are variable in repeat number.
- An individual’s genome changes over time, in specific sets of cells, as new mutations occur. These changes can be important if they affect cell function. For example, a mutation in a gene that blocks cell division may result in reduced expression of that gene, and cancer can then occur.
Comparisons among sequenced genomes from prokaryotes and eukaryotes have revealed some of the evolutionary relationships among genes. Some genes are present in both prokaryotes and eukaryotes; others are only in eukaryotes; still others are only in animals, or only in vertebrates (FIGURE 12.12).
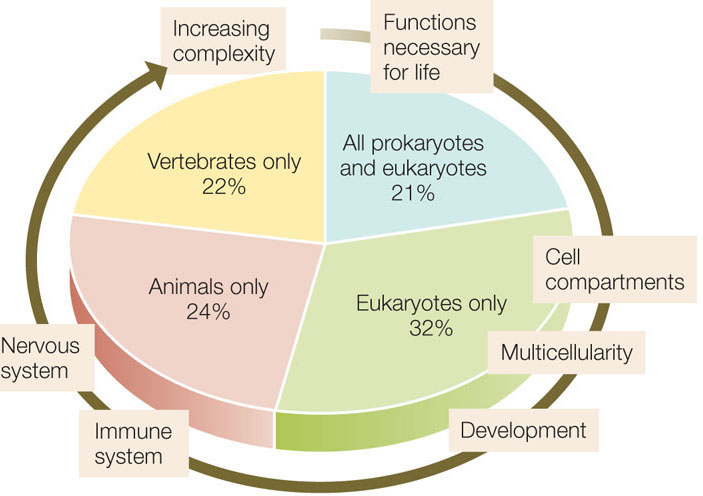
More comparative genomics is possible now that the genomes of other primates, including all the great apes, have been sequenced. Chimpanzees are our closest living relatives, sharing nearly 99 percent of our DNA sequence. Gorillas and orangutans are next closest, with genomes that are about 98 percent and 97 percent similar to ours. Researchers have identified about 500 protein-coding genes that have undergone accelerated evolution in humans, chimpanzees, and gorillas, including genes involved in hearing and brain development. Further analyses of these sequences may reveal genes that distinguish us from other apes and that “make humans human.”

Go to MEDIA CLIP 12.1 A Big Surprise from Genomics
PoL2e.com/mc12.1
Human genomics has potential benefits in medicine
Complex phenotypes are determined not by single genes but by multiple genes interacting with the environment. A single disease-causing allele, such as those associated with phenylketonuria and sickle-cell anemia (see Concept 9.3), does not exist for such common disorders as diabetes, heart disease, and Alzheimer’s disease. To understand the genetic bases of these diseases, biologists are now using rapid genotyping technologies to create “haplotype maps” that can be used to identify multiple genes involved in disease.
Haplotype Mapping
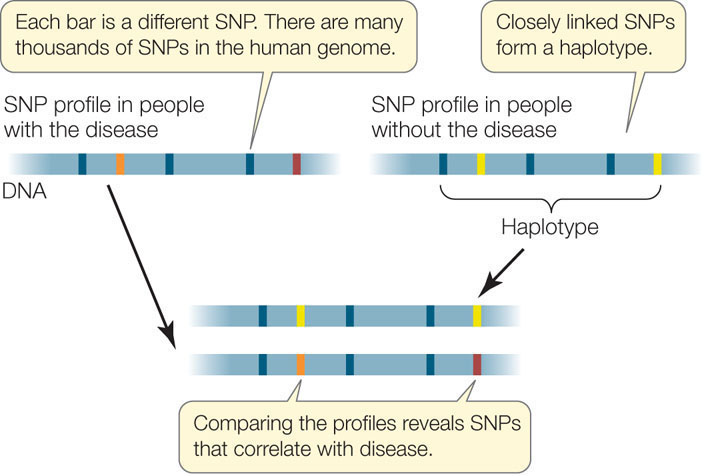
Haplotype maps are based on single nucleotide polymorphisms (SNPs)—DNA sequence variations that involve single nucleotides. SNPs (pronounced “snips”) arise as point mutations (see Concept 9.3). Because of these mutations, a single nucleotide in a homologous DNA sequence may vary among individuals or even between alleles in a single individual. Biologists use SNPs to create genetic maps of organisms, to classify organisms and species, and to identify individual organisms carrying specific alleles.
The SNPs that differ among individuals are not necessarily inherited as independent alleles. Rather, a set of SNPs that are close together on a chromosome is inherited as a unit (the SNPs are tightly linked). A piece of chromosome with a set of linked SNPs is called a haplotype. You can think of the haplotype as a sentence and the SNP as a word in the sentence. Analyses of haplotypes in humans from all over the world have thus far identified 500,000 common variations.
Genotyping Technology and Personal Genomics
New technologies are continually being developed to analyze thousands or millions of SNPs in the genomes of individuals. Such technologies include high-throughput sequencing methods and DNA microarrays, which depend on hybridization to identify specific SNPs.
A DNA microarray is a grid of microscopic spots of oligonucleotides (short DNA sequences) arrayed on a solid surface. It can be “probed” with a complex mixture of DNA or RNA; if the mixture contains a sequence that is complementary to one of the oligonucleotides, the sequence will hybridize to that spot. Colored fluorescent dyes are used to detect spots that hybridize with components of the probe mixture. The specific pattern of fluorescent dots reveals the haplotypes of the individual from whom the DNA came. For example, a microarray of 500,000 SNP-containing oligonucleotides has been used to analyze DNA from thousands of people to find out which SNPs are linked to genes associated with specific diseases. The aim is to identify particular alleles that contribute (along with particular alleles of other genes) to each complex disease (FIGURE 12.13). The amount of data from 500,000 SNPs and thousands of people with thousands of medical records is prodigious. With so much natural variation, statistical measures of association between a haplotype and a disease need to be very rigorous.
249
These association tests have revealed haplotypes or alleles that are associated with modestly increased risks for such diseases as breast cancer, diabetes, arthritis, obesity, and coronary heart disease. For example, 12 SNPs are associated with increased incidence of heart attacks, and if considered together, these SNPs can be used to identify individuals who are at increased risk. Indeed, the predictive value of this genetic test is greater than the widely used test for elevated blood cholesterol level. Private companies now offer to scan a human genome for SNP alleles, and the price for this service keeps getting lower. However, at this point it is unclear what a person without symptoms should do with the information, since multiple genes, environmental influences, and epigenetic effects all contribute to the development of these diseases.
Pharmacogenomics
Genetic variation can affect how an individual responds to a particular drug. For example, consider an enzyme in the liver that catalyzes the following reaction:
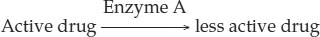
A mutation in the gene that encodes this enzyme may make the enzyme less active and reduce the rate at which the active drug is modified to a less active form. For a given dose of the drug, a person with the mutation would have more active drug in his or her bloodstream than a person without the mutation. So the dose of the drug needed for the same effect would be lower for this person.
Now consider a different case, in which a liver enzyme is needed to make the drug active:
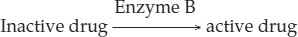
A person homozygous for a mutation in the gene encoding this enzyme would not be affected by the drug, since the activating enzyme is not present.
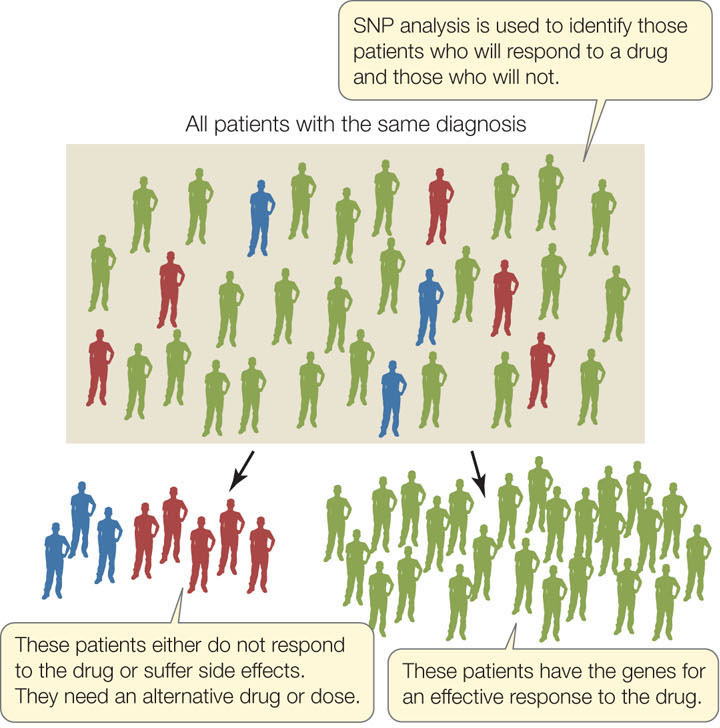
The study of how an individual’s genome affects his or her response to drugs or other agents is called pharmacogenomics. This type of analysis makes it possible to predict whether or not a drug will be effective. The objective is to personalize drug treatment so that a physician can know in advance whether an individual will benefit from a particular drug (FIGURE 12.14). This approach might also be used to reduce the incidence of adverse drug reactions in individuals who metabolize particular drugs slowly.
Proteomics
Comparisons of the proteomes of humans and other eukaryotic organisms have revealed a common set of proteins that can be categorized into groups (families) with similar amino acid sequences and similar functions. Forty-six percent of the yeast proteome, 43 percent of the worm proteome, and 61 percent of the fruit fly proteome are shared by the human proteome. Functional analyses indicate that this set of 1,300 proteins provides the basic metabolic functions of a eukaryotic cell—including glycolysis, the citric acid cycle, membrane transport, protein synthesis and targeting, and DNA replication. Of course, these are not the only human proteins. There are many more, which presumably distinguish us as human eukaryotic organisms.
250
There is considerable interest in using proteomics in the diagnosis of diseases. For diseases caused by a single gene, examining the single protein involved is possible (e.g., the hemoglobins; see Figure 10.2). But for more complex diseases such as diabetes and cancer, many genes and proteins may be involved, and the pattern of proteins made in a particular tissue at a certain time might indicate the presence or likelihood of the disease. Proteomic analyses of tissues (including blood) are being developed to provide early warnings of the presence of particular diseases before symptoms occur.
Metabolomics
There has been some progress in defining the human metabolome. A database created by David Wishart and colleagues at the University of Alberta contains more than 40,000 entries, including metabolic products of foods and drugs. The challenge now is to relate the levels of these substances to physiology. For example, high levels of glucose in the blood are associated with diabetes, and there may be patterns of metabolites that are diagnostic of other diseases. This could aid in early diagnosis and treatment.
DNA fingerprinting uses short tandem repeats
As noted in Concept 12.3, short tandem repeats (STRs) are blocks of 1–5 bp that can be repeated up to 100 times at particular locations on chromosomes. Since the number of repeats can vary widely, there are usually numerous alleles for a particular STR. For example, at a particular location on human chromosome 15 there might be an STR of AGG. An individual might inherit an allele with six copies of the repeat (AGGAG-GAGGAGGAGGAGG) from her mother and an allele with two copies (AGGAGG) from her father (FIGURE 12.15A). PCR can be used to amplify DNA fragments containing these repeat sequences, and the number of copies of the repeat can be determined by sizing the DNA fragments.
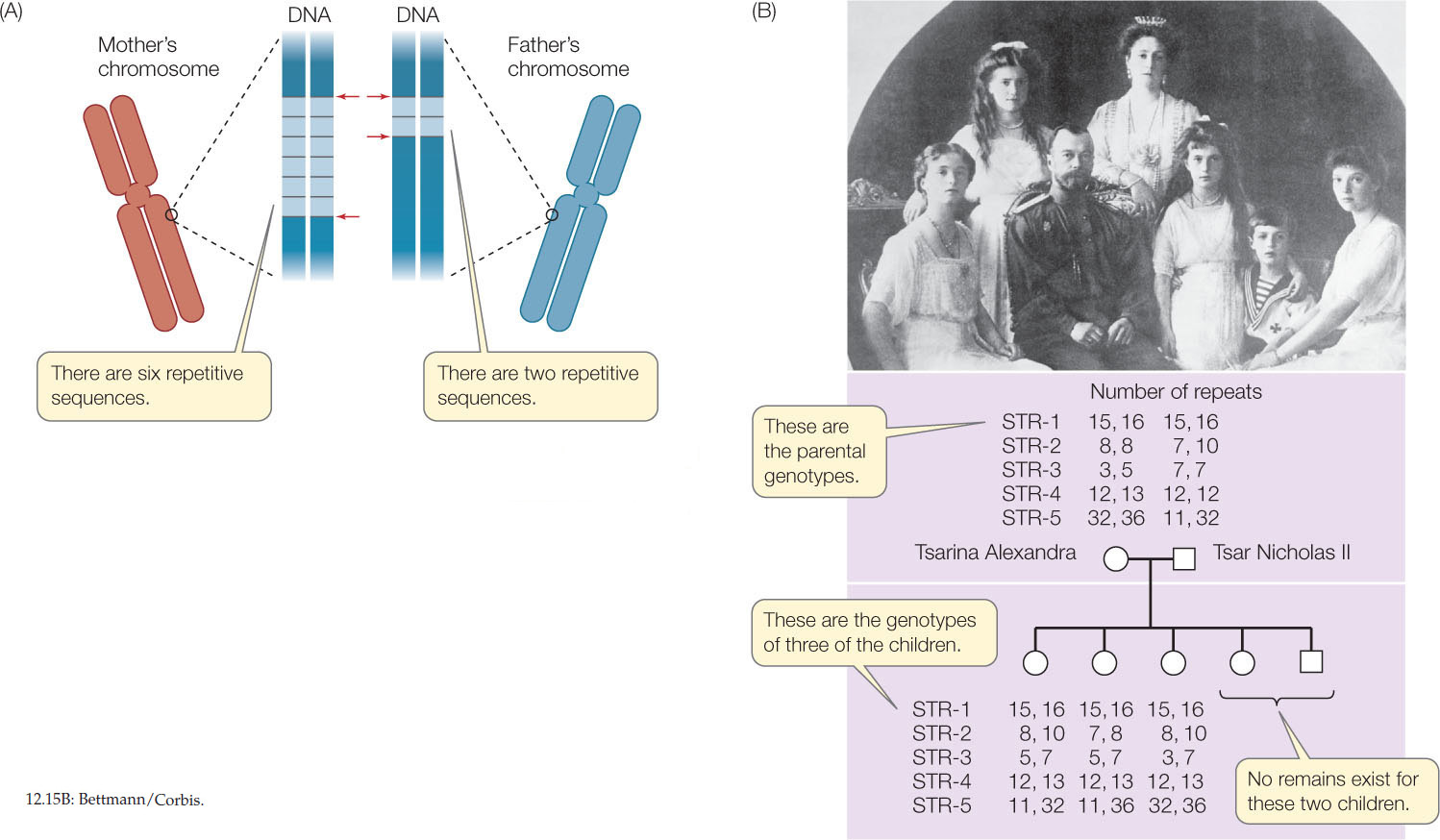
DNA fingerprinting refers to a group of techniques used to identify particular individuals by their DNA; the most common of these techniques involves STR analysis. When several different STR loci are analyzed, an individual’s unique pattern becomes apparent. The U.S. Federal Bureau of Investigation uses 13 STR loci in its Combined DNA Index System (CODIS) database.
DNA fingerprinting is used to resolve questions of paternity, and in forensics (crime investigations) to identify criminals. It also has other uses—for example, it can help in analyses of historical events. In 1918 the Russian tsar Nicholas II and his wife and five children were killed during the Communist revolution. A report that the bodies had been burned was not questioned until 1991, when a shallow grave with several skeletons was discovered a few miles from the presumed execution site. The remains were from a man, a woman, and three female children, and STR analysis indicated that they were all related to one another (FIGURE 12.15B). These STR patterns were also related to living descendants of the tsar. The accuracy and specificity of this method gave historical and cultural closure to a major event of the twentieth century.
Genome sequencing is at the leading edge of medicine
With the rapid development of better and better ways to sequence genomes cheaply, we are on the verge of a new era in medicine. The genomes of random cells of people, as well as cells from diseases such as cancers, are being sequenced in two ways:
- Sequencing of the entire genome
- Sequencing of protein-coding exons only
251
APPLY THE CONCEPT: The human genome sequence has many applications
It is the year 2025. You are taking care of a patient who is worried that he may have an early stage of kidney cancer. His mother died from this disease.
- Assume that the SNPs linked to genes involved in the development of this type of cancer have been identified. How would you determine if this man has a genetic predisposition for developing kidney cancer? Explain how you would do the analysis.
- How might you develop a metabolomic profile for kidney cancer and then use it to determine whether your patient has kidney cancer?
- If the patient is diagnosed with the cancer, how would you use pharmacogenomics to choose the right medications to treat his tumor?
The information these methods provide goes far beyond what SNP and STR genotyping can do. For example:
- The genome sequence of a tumor included mutations in specific genes that drive tumor formation. The proteins encoded by these genes are targets for specific therapy for that tumor.
- The genome sequence of an individual with a genetic disease has led to the identification of the causative gene.
CHECKpoint CONCEPT 12.4
- The average human gene spans 27,000 bp. The average human polypeptide has 300 amino acids. Explain.
- What is a haplotype with regard to an STR? How can haplotypes be used to relate DNA to a phenotype?
- A person has a rare allele for an STR (STR-1) that has a frequency in the population of 1 percent (0.01). The same person has allele frequencies for other STRs as follows: STR-2, 0.005; STR-3, 0.01; STR-4, 0.05; STR-5, 0.01. What is the probability that an individual will have all of these alleles? What does this mean in terms of identifying an individual with this genotype?
Question 12.2
What does genome sequencing reveal about dogs and other animals?
ANSWER In the opening story of this chapter we described how genome sequencing is being applied to breeds of dogs. For example, high-throughput sequencing methods (Concept 12.1) have allowed biologists to collect data on genes that control body size. This has led to the identification of a SNP (Concept 12.4) in the gene for Insulin-like growth factor 1 (IGF-1) that is important in determining size. Large breeds have an allele that encodes an active IGF-1, and small breeds have a different allele that encodes a less active version of the protein. In humans, IGF-1 mediates the overall effects of growth hormone, and people with a mutation in the IGF-1 gene have short stature.
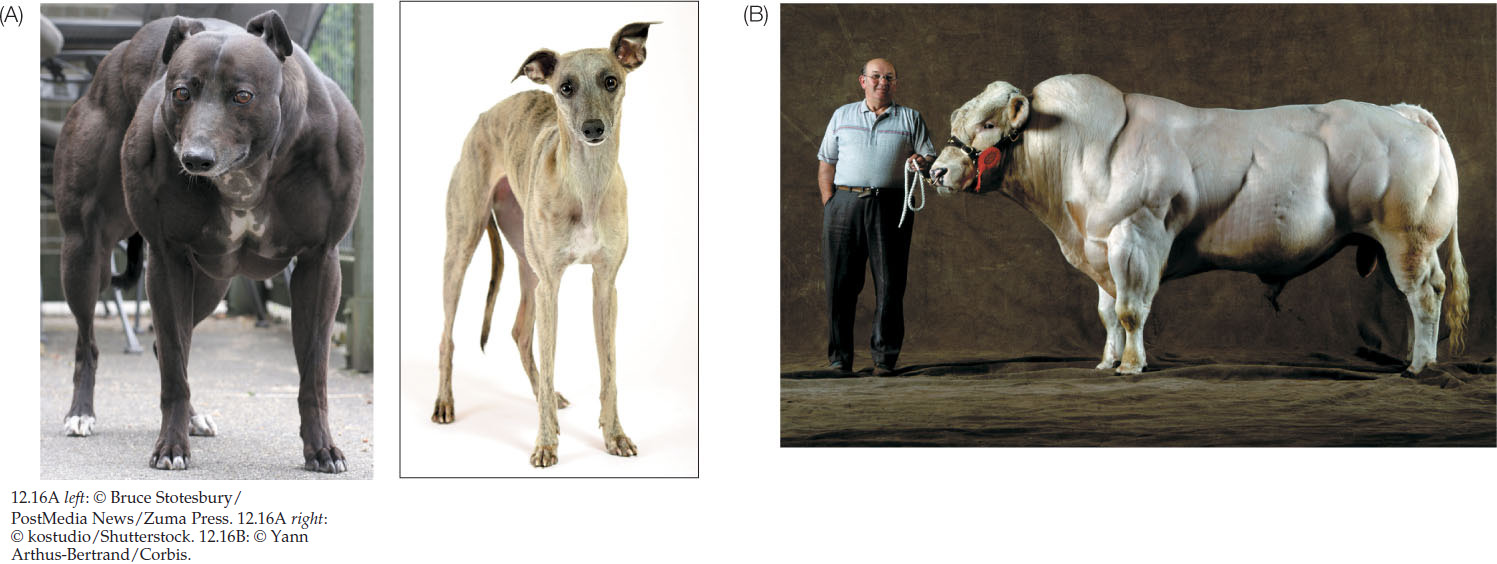
Another gene important to phenotypic variation is found in whippets, sleek dogs that run fast and are often raced. A mutation in the gene for myostatin, a protein that inhibits the overdevelopment of muscles, results in a whippet that is more muscular and runs faster (FIGURE 12.16A). Comparative genomics shows that this gene is important in other animals as well. In Belgian Blue cattle, individuals homozygous for a particular SNP in the myostatin gene have huge muscles (FIGURE 12.16B). There is interest in applying this knowledge to humans. For example, in muscular dystrophy the skeletal muscles waste away, and blocking myostatin could be useful in keeping muscles robust. Athletes anxious to have bulkier muscles have also been focusing on this gene and its protein product.
252
Inevitably, some scientists have set up companies to test dogs for genetic variations using DNA supplied by dog owners and breeders. The black dog rescued from the pound that looks like a Labrador retriever may turn out to be a German pointer. Some traditional breeders frown on this practice, but others say it will bring more joy (and prestige) to owners.