Concept 13.1: Recombinant DNA Can Be Made in the Laboratory
Biotechnology began with the use of organisms with genetic capabilities that occur in nature. For example, existing biochemical pathways in yeast are used to make alcohol, and as noted in the opening story, bacteria can be grown in large quantities to make acetone and other industrial chemicals. More recently, it has become possible to genetically modify organisms with genes from other, distantly related organisms, to create new combinations of genes that would not otherwise occur in the same cell. This technology involves the use of recombinant DNA: a single DNA molecule containing DNA sequences from two or more sources. Before the invention of PCR (polymerase chain reaction; see Concept 13.3 and also Concept 9.2), biologists relied on natural molecules and processes to manipulate DNA in the laboratory. Three key tools were, and still are, widely used:
- Restriction enzymes for cutting DNA into pieces (fragments) that can be manipulated
- Gel electrophoresis for the analysis and purification of DNA fragments
- DNA ligase for joining DNA fragments together in novel combinations
Restriction enzymes cleave DNA at specific sequences
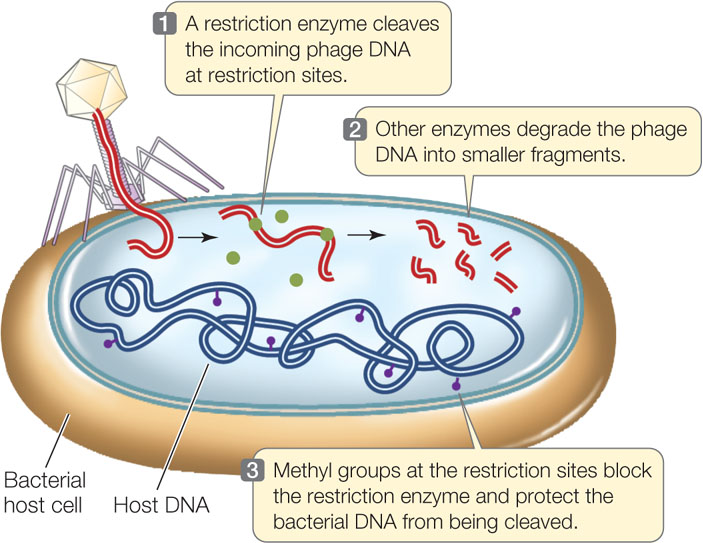
All organisms, including bacteria, must have ways of dealing with their enemies. As we described in Concept 9.1, bacteria are attacked by viruses called bacteriophage (or phage, for short). These viruses inject their genetic material into the host cells and turn them into virus-producing factories, eventually killing the cells. Some bacteria defend themselves against such invasions by producing restriction enzymes (also known as restriction endonucleases), which cut double-stranded DNA molecules—such as those injected by bacteriophage—into smaller, noninfectious fragments (FIGURE 13.1). These enzymes break the bonds of the DNA backbone between the 3′ hydroxyl group of one nucleotide and the 5′ phosphate group of the next nucleotide:

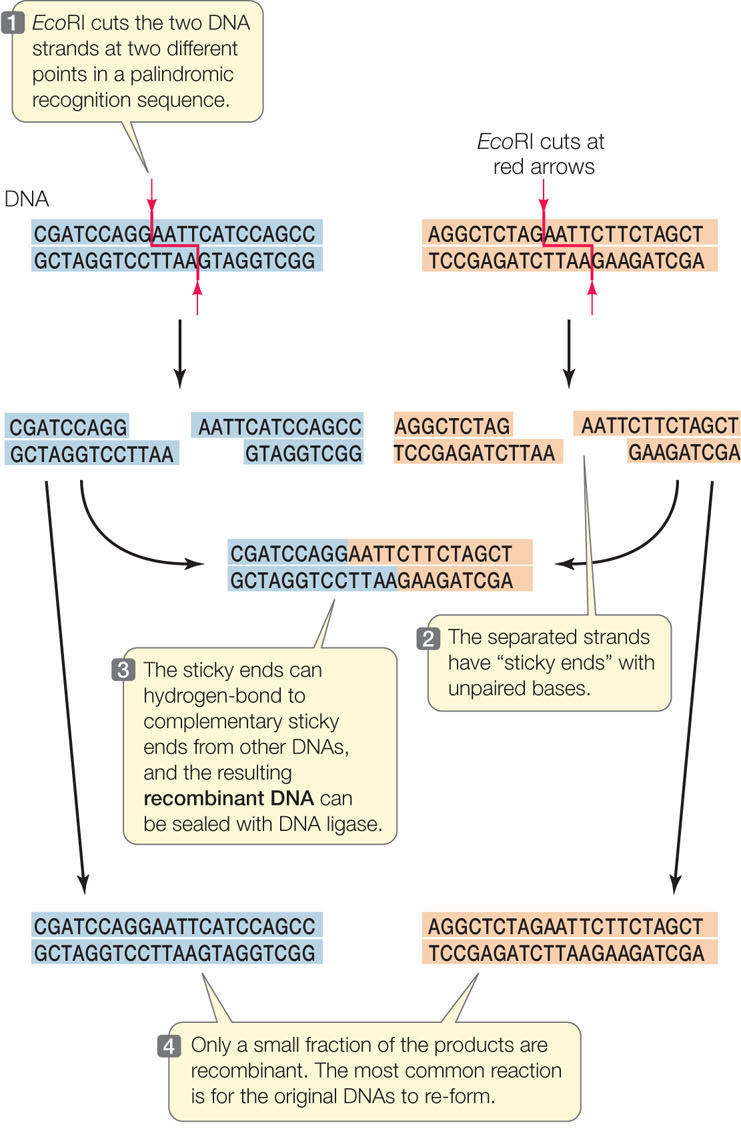
There are many different restriction enzymes, each of which cleaves DNA at a specific sequence of bases called a restriction site or recognition sequence. Hundreds of these enzymes have been purified from various microorganisms and can be used to cut DNA in the laboratory (by setting up a “restriction digest”). Most restriction sites are four to six base pairs (bp) long, and restriction enzymes catalyze hydrolysis of both strands of DNA. For example, below are three 6-bp sequences, each of which is recognized by a different restriction enzyme. The enzymes cleave the DNA at the sites indicated by the red arrows:

The names of these enzymes reflect their organism of origin. EcoRI, for example, was found in E. coli, and BamHI comes from the bacterium Bacillus amyloliquifaciens. Note that in each of these restriction sites both strands have the same sequence when read from their 5′ ends. This is similar to the word “race-car,” which is the same when read in either direction. A word that is the same when read in either direction is called a palindrome. Restriction enzymes have two identical active sites on two subunits, which cleave the two strands simultaneously:

255
Note also that these enzymes cut the two DNA strands in such a way that there will be a short sequence of single-stranded DNA at each cut end. Many restriction enzymes create these single-stranded overhangs, which are referred to as “sticky ends” because they are able to form hydrogen bonds with complementary sequences on other DNA molecules. Other restriction enzymes make cuts directly opposite one another on the two DNA strands, creating “blunt ends.”
As shown in Figure 13.1, the restriction enzymes may cleave host bacterial DNA. To prevent cleavage, a “stop sign” in the form of a methyl (—CH3) group can be placed on restriction sites. This process involves specific DNA methyltransferases. The restriction enzymes do not recognize or cut the methylated restriction sites in the host’s DNA. But unmethylated phage DNA is efficiently recognized and cleaved.
The EcoRI restriction site recognition sequence occurs, on average, about once in every 4,000 bp in a typical prokaryotic genome, or about once per four prokaryotic genes. So EcoRI can chop a large piece of DNA into smaller pieces containing, on average, just a few genes. When EcoRI is used in the laboratory to cut a small genome such as that of a virus with, say, 50,000 bp, only a few fragments are obtained. For a huge eukaryotic chromosome with tens of millions of bp, a very large number of fragments are created.
Of course, “on average” does not mean that the enzyme cuts all stretches of DNA at regular intervals. For example, the EcoRI restriction site does not occur even once in the 40,000 bp of the T7 phage genome—a fact that is crucial to the survival of this virus, since its host is E. coli. Fortunately for E. coli, the EcoRI restriction site does appear in the DNA of other bacteriophage.

Go to MEDIA CLIP 13.1 Striking Views of Recombinant DNA Being Made
PoL2e.com/mc13.1
Gel electrophoresis separates DNA fragments
After a sample of DNA has been cut with a restriction enzyme, the fragments can be separated from each other to determine the number of fragments and the size (in bp) of each fragment. In this way an individual fragment can be identified, and it can then be purified for further analysis or for use in an experiment.
A convenient way to separate or purify DNA fragments is by gel electrophoresis. Samples containing the fragments are placed in wells at one end of a semisolid gel (usually made of agarose or polyacrylamide polymers), and an electric field is applied to the gel (FIGURE 13.2). Because of its phosphate groups, DNA is negatively charged at neutral pH. Therefore, because opposite charges attract, the DNA fragments move through the gel toward the positive end of the field. Because the spaces between the polymers of the gel are small, small DNA molecules can move through the gel faster than larger ones. Thus DNA fragments of different sizes separate from one another and can be detected with a dye. This gives us three types of information about a DNA sample:
- The number of fragments. The number of fragments produced by digestion of a DNA sample with a given restriction enzyme depends on how many times that enzyme’s restriction site occurs in the sample. Thus gel electrophoresis can provide some information about the presence of specific DNA sequences in the DNA sample.
- The sizes of the fragments. DNA fragments of known size are often placed in one well of the gel to provide a standard for comparison. This tells us how large the DNA fragments in the other wells are. By comparing the fragment sizes obtained with two or more restriction enzymes, the locations of their recognition sequences relative to one another can be worked out (mapped).
- The relative abundance of a fragment. In many experiments, the investigator is interested in how much DNA is present. The relative intensity of a band produced by a specific fragment can indicate the amount of that fragment.
RESEARCH TOOLS

256
APPLY THE CONCEPT: Recombinant DNA can be made in the laboratory
The specificity of restriction enzyme recognition can be used to detect mutations. For example, the enzyme MstII cuts DNA at CCTNAGG, where N is any base. Around the sixth codon in the β-globin gene is the sequence CCTGAGG. There are two additional MstII sites on either side of the sixth codon, such that when MstII is used to cut human DNA in this region, two fragments of 1.15 and 0.20 kilobases are obtained (1 kilobase = 1,000 bp). The sickle allele of the β-globin gene causes sickle-cell anemia when it occurs in the homozygous state. In this allele, the sequence around the sixth codon is mutated to CCTGTGG.
- Identify the point mutation that led to the sickle allele.
- What fragment(s) would result when MstII is used to cut DNA with the sickle allele?
- Sketch the patterns of cuts with normal and sickle alleles on a gel.
- Could an individual have both patterns? Explain.
- How can this information be used to make a DNA test for these alleles?
After separation on the gel, a slice of gel containing the desired DNA fragment (identified by its size) can be cut out and then be purified by one of a variety of methods. This fragment can then be analyzed to determine its sequence or used to make recombinant DNA.

Go to ANIMATED TUTORIAL 13.1 Separating Fragments of DNA by Gel Electrophoresis
PoL2e.com/at13.1
LINK
Determining and then comparing DNA sequences of the same gene from different species provides information about evolutionary relationships; see Concepts 16.2 and 16.3
257
Recombinant DNA can be made from DNA fragments
Another enzyme that is involved in DNA metabolism in cells is DNA ligase, which catalyzes the joining of DNA fragments by making phosphodiester bonds between them. This is the enzyme that joins Okazaki fragments during DNA replication (see Concept 9.2). With restriction enzymes (which break bonds) and DNA ligase (which makes bonds), scientists can cut DNA into fragments and then join the fragments together in new combinations as recombinant DNA. As shown in FIGURE 13.3, two fragments with complementary sticky ends first join by hydrogen bonding, and then the DNA ligase forms a phosphodiester bond in each strand, making a single intact DNA molecule.
In the early 1970s, Stanley Cohen and Herbert Boyer wondered whether recombinant DNA could be a functional carrier of genetic information. They used restriction enzymes to cut sequences from two E. coli plasmids (small circular DNAs; see Concept 8.4) containing different antibiotic resistance genes. Then they used DNA ligase to join the fragments together. The resulting plasmid, when inserted into new E. coli cells, gave those cells resistance to both antibiotics (FIGURE 13.4). A new era of biotechnology was born.
Investigation
HYPOTHESIS
Biologically functional recombinant plasmids can be made in the laboratory.
METHOD
E. coli plasmids carrying a gene for resistance to either the antibiotic kanamycin (kanr) or tetracycline (tetr) are cut with a restriction enzyme.

CONCLUSION
Two DNA fragments with different genes can be joined to make a recombinant DNA molecule, and the resulting DNA is functional.
ANALYZE THE DATA
Two plasmids were used in this study: pSC101 had a gene for resistance to tetracycline and pSC102 had a gene for resistance to kanamycin. Equal quantities of the plasmids—either intact, cut with EcoRl, or cut with EcoRl and then sealed with DNA ligase—were mixed and incubated with antibiotic-sensitive E. coli. The E. coli were then grown on various combinations of the antibiotics. Here are the results:

- Did treatment with EcoRI affect the transformation efficiency? Explain.
- Did treatment with DNA ligase affect the transformation efficiency of each cut plasmid? Which quantitative data support your answer?
- How did doubly antibiotic-resistant bacteria arise in the “none” treatment? (Hint: see Concept 9.3.)
- Did the EcoRl followed by ligase treatment increase the appearance of doubly antibiotic-resistant bacteria? What data support your answer?
Go to LaunchPad for discussion and relevant links for all INVESTIGATION figures.
aS. N. Cohen et al. 1973. Proceedings of the National Academy of Sciences USA 70: 3240–3244.
258
CHECKpoint CONCEPT 13.1
- Using diagrams of the chemical structure of DNA (see Concepts 3.1 and 9.2), compare the actions of a restriction enzyme and DNA ligase.
- How is recombinant DNA technology different from genetic recombination that occurs in meiosis?
- A DNA molecule of 12,000 bp (12 kilobase, or kb) is cut by restriction enzymes and analyzed by gel electrophoresis as shown in the table. The 2-kb band from the double digest (Enzymes A + B) is twice as intense as the 2-kb bands from the single digests.

- Sketch the results of the three cuts on an electrophoresis gel.
- Indicate on a linear map where each enzyme cuts the DNA.
- DNA from two different sources is cut with two different restriction enzymes. Fragments from the two sources are then joined together. DNA from source A is cut with SpeI. Which of the four restriction enzymes shown below SpeI could be used to cut DNA from source B, such that it could join to a fragment from source A?

With the tools described in this concept—restriction enzymes, gel electrophoresis, and DNA ligase—scientists can cut and rejoin different DNA molecules from any and all sources, including artificially synthesized DNA sequences. In the next concept we will examine some of the ways these recombinant DNA molecules are used.