Concept 16.1: All of Life Is Connected through Its Evolutionary History
The sequencing of complete genomes from many diverse species has confirmed what biologists have long suspected: all of life is related through a common ancestor. The common ancestry of life explains why the general principles of biology apply to all organisms. Thus we can learn much about how the human genome works by studying the biology of model organisms because we share a common evolutionary history with those organisms. The evolutionary history of these relationships is known as phylogeny, and a phylogenetic tree is a diagrammatic reconstruction of that history.
Phylogenetic trees are commonly used to depict the evolutionary history of species, populations, and genes. For many years such trees have been constructed based on physical structures, behaviors, and biochemical attributes. Now, as genomes are sequenced for more and more organisms, biologists are able to reconstruct the history of life in ever greater detail.
In Chapter 15 we discussed why we expect populations of organisms to evolve over time. Such a series of ancestor and descendant populations forms a lineage, which we can depict as a line drawn on a time axis:

What happens when a single lineage divides into two? For example, a geographic barrier (such as a new mountain range) may divide an ancestral population into two descendant populations that no longer interact with one another. We depict such an event as a split, or node, in a phylogenetic tree. Each of the descendant populations gives rise to a new lineage, and as these independent lineages evolve, new traits arise in each:

As the lineages continue to split over time, this history can be represented in the form of a branching tree that can be used to trace the evolutionary relationships from the ancient common ancestor of a group of species, through the various lineage splits, up to the present populations of the organisms:
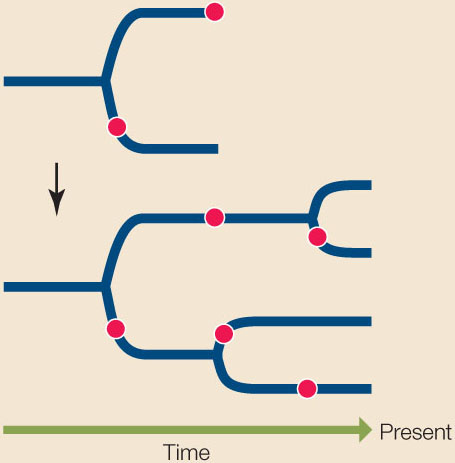
A phylogenetic tree may portray the evolutionary history of all life forms. Phylogenetic trees can also depict the history of a major evolutionary group (such as the insects) or of a much smaller group of closely related species. In some cases, phylogenetic trees are used to show the history of individuals, populations, or genes within a species. The common ancestor of all the organisms in the tree forms the root of the tree. The depictions of phylogenetic trees in this book are rooted at the left, with time flowing from left (earliest) to right (most recent):

The timing of splitting events in lineages is shown by the position of nodes on a time axis. These splits represent events where one lineage diverged into two, such as a speciation event (for a tree of species), a gene duplication event (for a tree of genes), or a transmission event (for a tree of viral lineages transmitted through a host population). The time axis may have an explicit scale, or it may simply show the relative timing of divergence events.
327
In this book’s illustrations, the order in which nodes are placed along the horizontal (time) axis has meaning, but the vertical distance between the branches does not. Vertical distances have been adjusted for legibility and clarity of presentation; they do not correlate with the degree of similarity or difference among groups. Note too that lineages can be rotated around nodes in the tree, so the vertical order of lineages is also largely arbitrary:

Any group of species that we designate with a name is a taxon (plural taxa). Examples of familiar taxa include humans, primates, mammals, and vertebrates; in this series, each taxon is also a member of the next, more inclusive taxon. Any taxon that consists of all the evolutionary descendants of a common ancestor is called a clade. Clades can be identified by picking any point on a phylogenetic tree and from that point tracing all the descendant lineages to the tips of the terminal branches (FIGURE 16.1). Two species that are each other’s closest relatives are called sister species. Similarly, any two clades that are each other’s closest relatives are sister clades.

Before the 1980s, phylogenetic trees tended to be seen only in the literature on evolutionary biology, especially in the area of systematics—the study and classification of biodiversity. But almost every journal in the life sciences published during the last few years contains phylogenetic trees. Trees are widely used in molecular biology, biomedicine, physiology, behavior, ecology, and virtually all other fields of biology. Why have phylogenetic studies become so widespread?
Phylogenetic trees are the basis of comparative biology
In biology we study life at all levels of organization—from genes, cells, organisms, populations, and species to the major divisions of life. In most cases, however, no individual gene or organism (or other unit of study) is exactly like any other gene or organism that we investigate.
Consider the individuals in your biology class. We recognize each person as an individual human, but we know that no two are exactly alike. If we knew everyone’s family tree in detail, the genetic similarity of any pair of students would be more predictable. We would find that more closely related students have many more traits in common (from the color of their hair to their susceptibility or resistance to diseases). Likewise, biologists use phylogenies to make comparisons and predictions about shared traits across genes, populations, and species.
The evolutionary relationships among species, as represented in the tree of life, form the basis for biological classification. Biologists estimate that there are tens of millions of species on Earth. So far, however, only about 1.8 million species have been classified—that is, formally described and named. New species are being discovered all the time and phylogenetic analyses are constantly reviewed and revised, so our knowledge of the tree of life is far from complete. Yet knowledge of evolutionary relationships is essential for making comparisons in biology, so biologists build phylogenies for groups of interest as the need arises. The tree of life’s evolutionary framework allows us to make many predictions about the behavior, ecology, physiology, genetics, and morphology of species that have not yet been studied in detail.
When biologists compare species, they observe traits that differ within the group of interest and try to understand when these traits evolved. In many cases, investigators are interested in how the evolution of a trait relates to environmental conditions or selective pressures. For instance, scientists have used phylogenetic analyses to discover changes in the genome of human immunodeficiency viruses that result in resistance to particular drug treatments. The association of a particular genetic change in HIV with a particular treatment provides a hypothesis about the evolution of resistance that can be tested experimentally.
Any features shared by two or more species that have been inherited from a common ancestor are said to be homologous. Homologous features may be any heritable traits, including DNA sequences, protein structures, anatomical structures, and even some behavior patterns. For example, all living vertebrates have a vertebral column, as did the ancestral vertebrate. Therefore the vertebral column is judged to be homologous in all vertebrates.
328
Derived traits provide evidence of evolutionary relationships
In tracing the evolution of a character, biologists distinguish between ancestral and derived traits. Each character of an organism evolves from one condition, called the ancestral trait, to another condition, called the derived trait.
Derived traits that are shared among a group of organisms and are also viewed as evidence of the common ancestry of the group are called synapomorphies (syn, “shared”; apo, “derived”; morph, “form,” referring to the “form” of a trait). Thus the vertebral column is considered a synapomorphy—a shared, derived trait—of the vertebrates. (The ancestral trait was an undivided supporting rod.)
Not all similar traits are evidence of relatedness. Similar traits in unrelated groups of organisms can develop for either of the following reasons:
- Superficially similar traits may evolve independently in different lineages, a phenomenon called convergent evolution. For example, although the wing bones of bats and birds are homologous, having been inherited from a common tetrapod ancestor, the wings of bats and birds are not homologous because they evolved independently from the forelimbs of different nonflying ancestors (FIGURE 16.2).
 Figure 16.2: The Bones Are Homologous, the Wings Are Not The supporting bone structures of both bat wings and bird wings are derived from a common tetrapod (four-limbed) ancestor and are thus homologous. However, the wings themselves—an adaptation for flight—evolved independently in the two groups.
Figure 16.2: The Bones Are Homologous, the Wings Are Not The supporting bone structures of both bat wings and bird wings are derived from a common tetrapod (four-limbed) ancestor and are thus homologous. However, the wings themselves—an adaptation for flight—evolved independently in the two groups. - A character may revert from a derived state back to an ancestral state in an event called an evolutionary reversal. For example, the derived limbs of terrestrial tetrapods evolved from the ancestral fins of their aquatic ancestors. Then, within the mammals, the ancestors of modern cetaceans (whales and dolphins) returned to the ocean, and cetacean limbs evolved to once again resemble their ancestral state—fins. The superficial similarity of cetacean and fish fins does not suggest a close relationship between these groups. Instead, the similarity arises from evolutionary reversal.
Similar traits generated by convergent evolution and evolutionary reversals are called homoplastic traits or homoplasies.

Go to MEDIA CLIP 16.1 Morphing Arachnids
PoL2e.com/mc16.1
A particular trait may be ancestral or derived, depending on our point of reference. For example, all birds have feathers. We infer from this that feathers (which are highly modified scales) were present in the common ancestor of modern birds. Therefore we consider the presence of feathers to be an ancestral trait for any particular group of modern birds, such as the songbirds. However, feathers are not present in any other living animals. In reconstructing a phylogeny of all living vertebrates, the presence of feathers is a derived trait found only among birds, and thus is a synapomorphy of the birds.
CHECKpoint CONCEPT 16.1
 What biological processes can be represented in a phylogenetic tree?
What biological processes can be represented in a phylogenetic tree?
Why is it important to consider only homologous characters in constructing phylogenetic trees?
What are some reasons that similar traits might arise independently in species that are only distantly related? Can you think of examples among familiar organisms?
Phylogenetic analyses of evolutionary history have become increasingly important to many types of biological research in recent years, and they are the basis for the comparative nature of biology. For the most part, however, evolutionary history cannot be observed directly. How, then, do biologists reconstruct the past?