Concept 16.2: Phylogeny Can Be Reconstructed from Traits of Organisms
To illustrate how a phylogenetic tree is constructed, consider the eight vertebrate animals listed in TABLE 16.1: lamprey, perch, salamander, lizard, crocodile, pigeon, mouse, and chimpanzee. We will initially assume that any given derived trait arose only once during the evolution of these animals (that is, there has been no convergent evolution), and that no derived traits were lost from any of the descendant groups (there has been no evolutionary reversal). For simplicity, we have selected traits that are either present (+) or absent (−).

329
In a phylogenetic study, the group of organisms of primary interest is called the ingroup. As a point of reference, an ingroup is compared with an outgroup: a species or group that is closely related to the ingroup but is known to be phylogenetically outside it. In other words, the root of the tree is located between the ingroup and the outgroup. Any trait that is present in both the ingroup and the outgroup must have evolved before the origin of the ingroup and thus must be ancestral for the ingroup. In contrast, traits that are present in only some members of the ingroup must be derived traits within that ingroup. As we will see in Chapter 23, a group of jawless fishes called the lampreys is thought to have separated from the lineage leading to the other vertebrates before the jaw arose. Therefore we have included the lamprey as the outgroup for our analysis. Because derived traits are traits acquired by other members of the vertebrate lineage after they diverged from the outgroup, any trait that is present in both the lamprey and the other vertebrates is judged to be ancestral.
We begin by noting that the chimpanzee and mouse share two traits—mammary glands and fur—that are absent in both the outgroup and in the other species of the ingroup. Therefore we infer that mammary glands and fur are derived traits that evolved in a common ancestor of chimpanzees and mice after that lineage separated from the lineages leading to the other vertebrates. These characters are synapomorphies that unite chimpanzees and mice (as well as all other mammals, although we have not included other mammalian species in this example). By the same reasoning, we can infer that the other shared derived traits are synapomorphies for the various groups in which they are expressed. For instance, keratinous scales are a synapomorphy of the lizard, crocodile, and pigeon.
Table 16.1 also tells us that, among the animals in our ingroup, the pigeon has a unique trait: the presence of feathers. Feathers are a synapomorphy of birds and their extinct relatives. However, because we only have one bird in this example, the presence of feathers provides no clues concerning relationships among these eight species of vertebrates. However, gizzards are found in both birds and crocodiles, so this trait is evidence of a close relationship between birds and crocodilians.
By combining information about the various synapomorphies, we can construct a phylogenetic tree. We infer from our information that mice and chimpanzees—the only two animals that share fur and mammary glands—share a more recent common ancestor with each other than they do with pigeons and crocodiles. Otherwise we would need to assume that the ancestors of pigeons and crocodiles also had fur and mammary glands but subsequently lost them. There is no need to make these additional assumptions.
FIGURE 16.3 shows a phylogenetic tree for the vertebrates in Table 16.1, based on the shared derived traits we examined. This particular tree was easy to construct because it is based on a very small sample of traits, and the derived traits we examined evolved only once and were never lost after they appeared. Had we included a snake in the group, our analysis would not have been as straightforward. We would have needed to examine additional characters to determine that snakes evolved from a group of lizards that had limbs. In fact, the analysis of many characters shows that snakes evolved from burrowing lizards that became adapted to a subterranean existence.

Go to ACTIVITY 16.1 Constructing a Phylogenetic Tree
PoL2e.com/ac16.1
Parsimony provides the simplest explanation for phylogenetic data
Typically, biologists construct phylogenetic trees using hundreds or thousands of traits. With larger data sets, we would expect to observe traits that have changed more than once, and thus would expect to see convergence and evolutionary reversal. How do we determine which traits are synapomorphies and which are homoplasies? One way is to invoke the principle of parsimony.
In its most general form, the parsimony principle states that the preferred explanation of observed data is the simplest explanation. Applying the principle of parsimony to the reconstruction of phylogenies entails minimizing the number of evolutionary changes that need to be assumed over all characters in all groups in the tree. In other words, the best hypothesis under the parsimony principle is one that requires the fewest homoplasies. This application of parsimony is a specific case of a general principle of reasoning called Occam’s razor: the best explanation is the one that best fits the data while making the fewest assumptions. More complicated explanations are accepted only when the evidence requires them. Phylogenetic trees represent our best estimates about evolutionary relationships, given our current knowledge. They are continually modified as additional evidence becomes available.
330
APPLY THE CONCEPT: Phylogeny can be reconstructed from traits of organisms
The matrix below supplies data for seven land plants and an outgroup (an aquatic plant known as a stonewort). Each trait is scored as either present (+) or absent (−) in each of the plants. Use this data matrix to reconstruct the phylogeny of land plants and answer the questions. See Activity 16.1 for help with constructing a phylogenetic tree.
- Which two of these taxa are most closely related?
- Plants that produce seeds are known as seed plants. What is the sister group to the seed plants among these taxa?
- Which two traits evolved along the same branch of your reconstructed phylogeny?
- Are there any homoplasies in your reconstructed phylogeny?
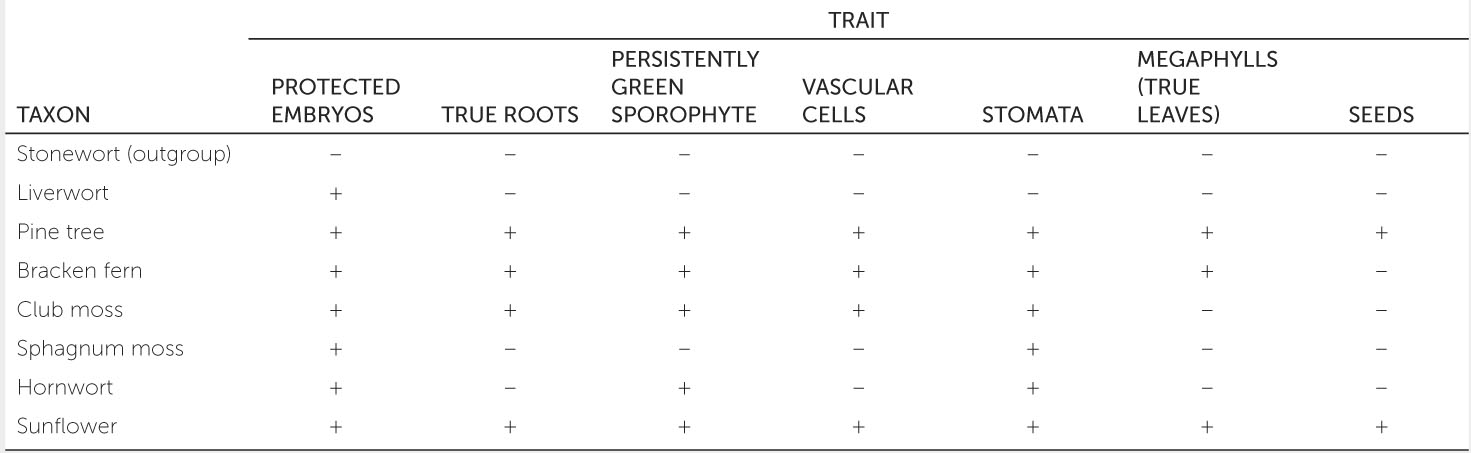
331
Phylogenies are reconstructed from many sources of data
Naturalists have constructed various forms of phylogenetic trees for more than 150 years. In fact, the only figure in the first edition of On the Origin of Species was a phylogenetic tree. Tree construction has been revolutionized, however, by the advent of computer software that allows us to consider far more data and analyze many more traits than could ever before be processed. Combining these advances in methodology with the massive comparative data sets being generated through studies of genomes, biologists are learning details about the tree of life at a remarkable pace (see Appendix A: The Tree of Life).
Any trait that is genetically determined, and therefore heritable, can be used in a phylogenetic analysis. Evolutionary relationships can be revealed through studies of morphology, development, the fossil record, behavioral traits, and molecular traits such as DNA and protein sequences. Let’s take a closer look at the types of data used in modern phylogenetic analyses.

Go to ANIMATED TUTORIAL 16.1 Phylogeny and Molecular Evolution
PoL2e.com/at16.1
Morphology
An important source of phylogenetic information is morphology: the presence, size, shape, and other attributes of body parts. Since living organisms have been observed, depicted, and studied for millennia, we have a wealth of recorded morphological data as well as extensive museum and herbarium collections of organisms whose traits can be measured. New technological tools, such as the electron microscope and computed tomography (CT) scans, enable systematists to examine and analyze the structures of organisms at much finer scales than was formerly possible.
Most species are described and known primarily by their morphology, and morphology still provides the most comprehensive data set available for many taxa. The morphological features that are important for phylogenetic analysis are often specific to a particular group. For example, the presence, development, shape, and size of various features of the skeletal system are important in vertebrate phylogeny, whereas floral structures are important for studying the relationships among flowering plants.
Morphological approaches to phylogenetic analysis have some limitations, however. Some taxa exhibit little morphological diversity, despite great species diversity. For example, the phylogeny of the leopard frogs of North and Central America would be difficult to infer from morphological differences alone, because the many species look very similar, despite important differences in their behavior and physiology. At the other extreme, few morphological traits can be compared across distantly related species (earthworms and mammals, for example). Furthermore, some morphological variation has an environmental (rather than a genetic) basis and so must be excluded from phylogenetic analyses. An accurate phylogenetic analysis often requires information beyond that supplied by morphology.
Development
Similarities in developmental patterns may reveal evolutionary relationships. Some organisms exhibit similarities in early developmental stages only. The larvae of marine creatures called sea squirts, for example, have a flexible gelatinous rod in the back—the notochord—that disappears as the larvae develop into adults. All vertebrate animals also have a notochord at some time during their development (FIGURE 16.4). This shared structure is one of the reasons for inferring that sea squirts are more closely related to vertebrates than would be suspected if only adult sea squirts were examined.

Link
For more on the role of developmental processes in evolution, see Concepts 14.4 and 14.5
Paleontology
The fossil record is another important source of information on evolutionary history. Fossils show us where and when organisms lived in the past and give us an idea of what they looked like. Fossils provide important evidence that helps us distinguish ancestral from derived traits. The fossil record can also reveal when lineages diverged and began their independent evolutionary histories. Furthermore, in groups with few species that have survived to the present, information on extinct species is often critical to an understanding of the large divergences among the surviving species. The fossil record has limitations, however. Few or no fossils have been found for some groups, and the fossil record for many groups is fragmentary.
Behavior
Some behavioral traits are culturally transmitted and others are genetically inherited. If a particular behavior is culturally transmitted, it may not accurately reflect evolutionary relationships (but may nonetheless reflect cultural connections). Many bird songs, for instance, are learned and may be inappropriate traits for phylogenetic analysis. Frog calls, however, are genetically determined and appear to be acceptable sources of information for reconstructing phylogenies.
Molecular Data
All heritable variation is encoded in DNA, and so the complete genome of an organism contains an enormous set of traits (the individual nucleotide bases of DNA) that can be used in phylogenetic analyses. In recent years, DNA sequences have become among the most widely used sources of data for constructing phylogenetic trees. Comparisons of nucleotide sequences are not limited to the DNA in the cell nucleus. Eukaryotes have genes in their mitochondria as well as in their nuclei. Plant cells also have genes in their chloroplasts. The chloroplast genome (cpDNA), which is used extensively in phylogenetic studies of plants, has changed slowly over evolutionary time, so it is often used to study relatively ancient phylogenetic relationships. Most animal mitochondrial DNA (mtDNA) has changed more rapidly, so mitochondrial genes are used to study evolutionary relationships among closely related animal species (the mitochondrial genes of plants evolve more slowly). Many nuclear gene sequences are also commonly analyzed, and now that entire genomes have been sequenced from many species, they too are used to construct phylogenetic trees. Information on gene products (such as the amino acid sequences of proteins) is also widely used for phylogenetic analyses.
332
Mathematical models expand the power of phylogenetic reconstruction
As biologists began to use DNA sequences to infer phylogenies in the 1970s and 1980s, they developed explicit mathematical models describing how DNA sequences change over time. These models account for multiple changes at a given position in a DNA sequence. They also take into account different rates of change at different positions in a gene, at different positions in a codon, and among different nucleotides. For example, transitions (changes between two purines or between two pyrimidines) are usually more likely than are transversions (changes between a purine and pyrimidine).
Mathematical models can be used to compute how a tree might evolve given the observed data. A maximum likelihood method will identify the tree that most likely produced the observed data, given the assumed model of evolutionary change. Maximum likelihood methods can be used for any kind of characters, but they are most often used with molecular data, for which explicit mathematical models of evolutionary change are easier to develop. The principal advantages to maximum likelihood analyses are that they incorporate more information about evolutionary change than do parsimony methods, and they are easier to treat in a statistical framework. The principal disadvantages are that they are computationally intensive and require explicit models of evolutionary change (which may not be available for some kinds of character change).
The accuracy of phylogenetic methods can be tested
How can we test the accuracy of phylogenetic methods? After all, phylogenetic trees represent reconstructions of past events, and many of these events occurred before any humans were around. To address this issue, biologists have conducted experiments both in living organisms and with computer simulations to test the effectiveness and accuracy of phylogenetic methods.
In one experiment designed to test the accuracy of phylogenetic analysis, a single viral culture of bacteriophage T7 was used as a starting point, and lineages were allowed to evolve from this ancestral virus in the laboratory (FIGURE 16.5). The initial culture was split into two separate lineages, one of which became the ingroup for analysis and the other of which became the outgroup for rooting the tree. The lineages in the ingroup were split in two after every 400 generations, and samples of the virus were saved for analysis at each branching point. The lineages were allowed to evolve until there were eight lineages in the ingroup. Mutagens were added to the viral cultures to increase the mutation rate so that the amount of change and the degree of homoplasy would be typical of the organisms analyzed in average phylogenetic analyses. The investigators then sequenced samples from the end points of the eight lineages, as well as from the ancestors at the branching points. They then gave the sequences from the end points of the lineages to other investigators to analyze, without revealing the known history of the lineages or the sequences of the ancestral viruses.
333
Investigation
HYPOTHESIS
A phylogenetic tree reconstructed from analysis of the DNA sequences of living organisms can accurately match the known evolutionary history of the organisms.
METHOD
In the laboratory, one group of investigators produced an experimental phylogeny of 9 viral lineages, enhancing the mutation rate to increase variation among the lineages.a

RESULTS
The true phylogeny and ancestral DNA sequences were accurately reconstructed solely from the DNA sequences of the viruses at the tips of the tree.
CONCLUSION
Phylogenetic analysis of DNA sequences can accurately reconstruct evolutionary history.
ANALYZE THE DATA
The full DNA sequences for the T7 strains in this experiment are thousands of nucleotides long. The nucleotides (“characters”) at 23 DNA positions are given in the table.b See Activity 16.1 for help with constructing a phylogenetic tree.
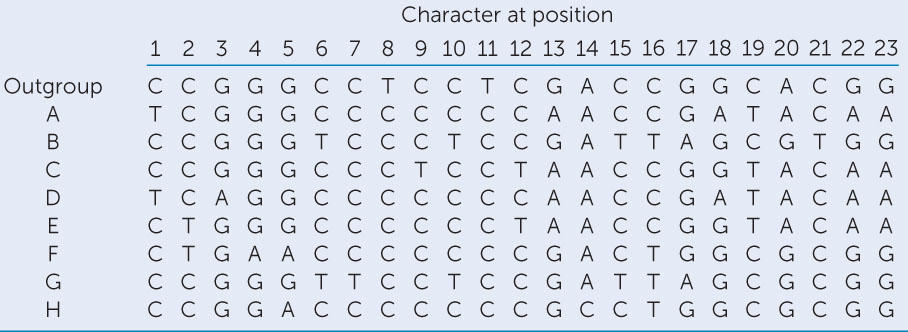
- Construct a phylogenetic tree from these DNA positions using the parsimony method. Use the outgroup to root your tree. Assume that all changes among nucleotides are equally likely.
- Using your tree, reconstruct the DNA sequences of the ancestral lineages.
Go to LaunchPad for discussion and relevant links for all INVESTIGATION figures.
aD. M. Hillis et al. 1992. Science 255: 589–295.
bJ. J. Bull et al. 1993. Evolution 47: 993–1007.
334
After the phylogenetic analysis was completed, the investigators asked two questions. Did phylogenetic methods reconstruct the known history correctly? And were the sequences of the ancestral viruses reconstructed accurately? The answer in both cases was yes. The branching order of the lineages was reconstructed exactly as it had occurred, more than 98 percent of the nucleotide positions of the ancestral viruses were reconstructed correctly, and 100 percent of the amino acid changes in the viral proteins were reconstructed correctly.
Go to ANIMATED TUTORIAL 16.2 Using Phylogenetic Analysis to Reconstruct Evolutionary History
PoL2e.com/at16.2
The experiment shown in Figure 16.5 demonstrated that phylogenetic analysis was accurate under the conditions tested, but it did not examine all possible conditions. Other experimental studies have taken other factors into account, such as the sensitivity of phylogenetic analysis to parallel selection and highly variable rates of evolutionary change. In addition, computer simulations based on evolutionary models have been used extensively to study the effectiveness of phylogenetic analysis. These studies have also confirmed the accuracy of phylogenetic methods and have been used to refine those methods and extend them to new applications.
CHECKpoint CONCEPT 16.2
- How is the parsimony principle used in reconstructing evolutionary history?
- Why is it useful to consider the entire life cycle when reconstructing an organism’s evolutionary history?
- What are some comparative advantages and disadvantages of morphological and molecular approaches for reconstructing phylogenetic trees?
- Contrast experimental and computer simulation approaches for testing the accuracy of phylogenetic reconstructions of evolutionary history. Can you think of some aspects of phylogenetic accuracy that might be more practical to test using computer simulation rather than experimental studies of viruses?
Why do biologists expend the time and effort necessary to reconstruct phylogenies? Information about the evolutionary relationships among organisms is a useful source of data for scientists investigating a wide variety of biological questions. Next we will describe how phylogenetic trees are used to answer questions about the past, and to predict and compare traits of organisms in the present.