4.1 MOLECULAR STRUCTURE OF PROTEINS
If you think of a protein as analogous to a word in the English language, then the amino acids are like letters. The comparison is not altogether fanciful, as there are about as many amino acids in proteins as letters in the alphabet, and the order of both amino acids and letters is important. For example, the word PROTEIN has the same letters as POINTER, but the two words have completely different meanings. Similarly, the exact order of amino acids in a protein makes a big difference because that order determines the protein’s shape and function.
4.1.1 Amino acids differ in their side chains.
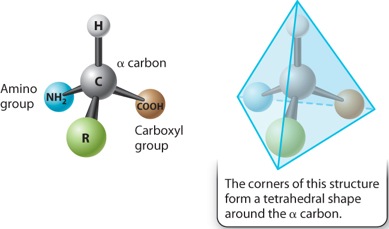
The general structure of an amino acid was discussed in Chapter 2 and is shown again in Figure 4.1. It consists of a central carbon atom, called the alpha (α) carbon, connected by covalent bonds to four different chemical groups: an amino group (–NH2 shown in blue), a carboxyl group (–COOH shown in brown), a hydrogen atom (–H shown in gray), and a side chain or R group (shown in green). The four covalent bonds from the α carbon are at equal angles. As a result, an amino acid forms a tetrahedron, a sort of pyramid with four triangular faces.
The R groups of the amino acids are also known as side chains, and they differ from one amino acid to the next. They are what make the “letters” of the amino acid “alphabet” distinct from one another. Just as letters differ in their shapes and sounds—vowels like E, I, and O and hard consonants like B, P, and T—amino acids differ in their chemical and physical properties.
The chemical structures of the 20 amino acids commonly found in proteins are shown in Figure 4.2. The side chains (shown in green) are chemically diverse and are grouped according to their properties, with a particular emphasis on whether they are hydrophobic or hydrophilic, or have special characteristics that might affect a protein’s structure. Within these broad categories are additional groupings based on whether they are nonpolar or polar, basic or acidic. These properties strongly influence how a polypeptide folds, and hence the three-dimensional shape of the protein.
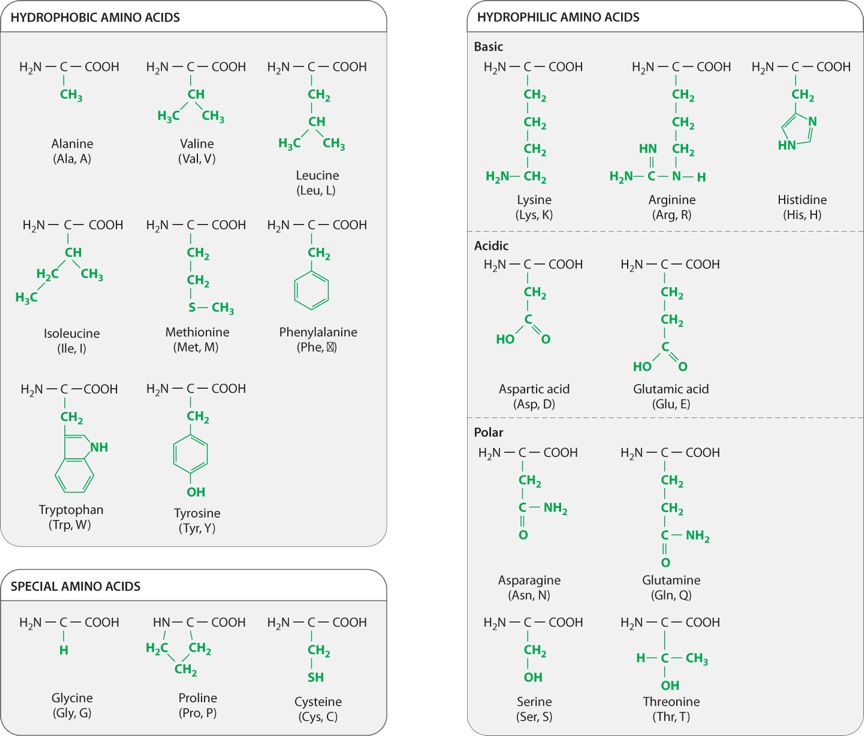
For example, as we saw in Chapter 2, hydrophobic molecules do not readily interact with water and do not easily form hydrogen bonds. Water molecules in the cell therefore tend to form hydrogen bonds with each other instead of with hydrophobic side chains. The aggregation of hydrophobic side chains is also stabilized by weak van der Waals forces (Chapter 2), in which asymmetries in electron distribution create temporary dipoles in the interacting molecules, which are then attracted to each other. The tendency for hydrophilic water molecules to interact with each other and for hydrophobic molecules to interact with each other is the very same tendency that forms oil droplets in water. This is also the reason why most hydrophobic amino acids tend to be buried in the interior of folded proteins, where they are kept away from water.
Amino acids with polar side chains have a permanent charge separation, in which one end of the side chain is slightly more negatively charged than the other. As we saw in Chapter 2, polar molecules are hydrophilic, and they tend to form hydrogen bonds with each other or with water molecules.
The side groups of the basic and acidic amino acids are strongly polar and are also hydrophilic. The side groups of basic amino acids tend to be positively charged at intracellular pH, and those of the acidic amino acids tend to be negatively charged. For this reason, basic and acidic side chains are usually located on the outside surface of the folded molecule. The charged groups can also form ionic bonds (negative with positive) with each other and with other charged molecules in the environment. This ability to bind another molecule of opposite charge is one important way that proteins can associate with each other or with other macromolecules such as DNA.
The properties of several amino acids are noteworthy because of their effect on protein structure. These amino acids include glycine, proline, and cysteine. Glycine is different from the other amino acids because its R group is hydrogen, exactly like the hydrogen on the other side, and therefore it is not asymmetric. All of the other amino acids have four different groups attached to the α carbon and are asymmetric. In addition, glycine is nonpolar and small enough to tuck into spaces where other R groups would not fit. The small size of glycine’s R group also allows for freer rotation around the C–N bond since its R group will not get in the way of the R groups of neighboring amino acids. Thus, glycine increases the flexibility of the polypeptide backbone, which can be important in the folding of the protein.
Proline is also distinctive, but for a different reason. Note how its R group is linked back to the amino group. This linkage creates a kink or bend in the polypeptide chain and restricts rotation of the C–N bond, thereby imposing constraints on protein folding in its vicinity, an effect the very opposite of glycine’s.
Cysteine makes a special contribution to protein folding through its –SH group. When two cysteine side chains in the same or different polypeptides come into proximity, they can react to form an S–S disulfide bond, which covalently joins the side chains. Such disulfide bonds form cross-bridges that can connect different parts of the same protein or even different proteins. This property contributes to the overall structure of single proteins or combinations of proteins.
4.1.2 Successive amino acids in proteins are connected by peptide bonds.
Amino acids are linked together to form proteins. Figure 4.3 shows how amino acids in a protein are bonded together. The bond formed between the two amino acids is a peptide bond, shown in red in Figure 4.3. In forming the peptide bond, the carboxyl group of one amino acid reacts with the amino group of the next amino acid in line, and a molecule of water is released. Note that in the resulting molecule, the R groups of each amino acid point in different directions.
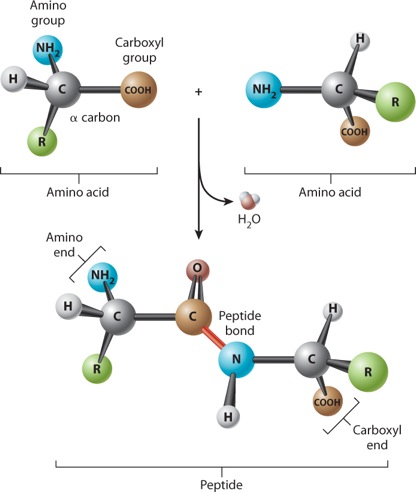
The C=O group in the peptide bond is known as a carbonyl group, and the N–H group is an amide group. Note in Figure 4.3 that these two groups are on either side of the peptide bond. This arrangement results in the delocalization of electrons, with the effect that the peptide bond has some of the characteristics of a double bond. The peptide bond is shorter than a single bond, for example, and it is not free to rotate like a single bond. The other bonds are free to rotate around their central axes.
Polymers of amino acids ranging from as few as two to many hundreds share a chemical feature common to individual amino acids: namely, that the ends are chemically distinct from each other. One end, shown at the left in Figure 4.3, has a free amino group; this is the amino end of the peptide. The other end has a free carboxyl group, which constitutes the carboxyl end of the molecule. More generally, a polymer of amino acids connected by peptide bonds is known as a polypeptide. Typical polypeptides produced in cells consist of a few hundred amino acids. In human cells, the shortest polypeptides are about 100 amino acids in length; the longest is the muscle protein titin, with 34,350 amino acids. The term protein is often used as a synonym for polypeptide, especially when the polypeptide chain has folded into a stable, three-dimensional conformation. Amino acids that are incorporated into a protein are often referred to as amino acid residues.
4.1.3 The sequence of amino acids dictates protein folding, which determines function.
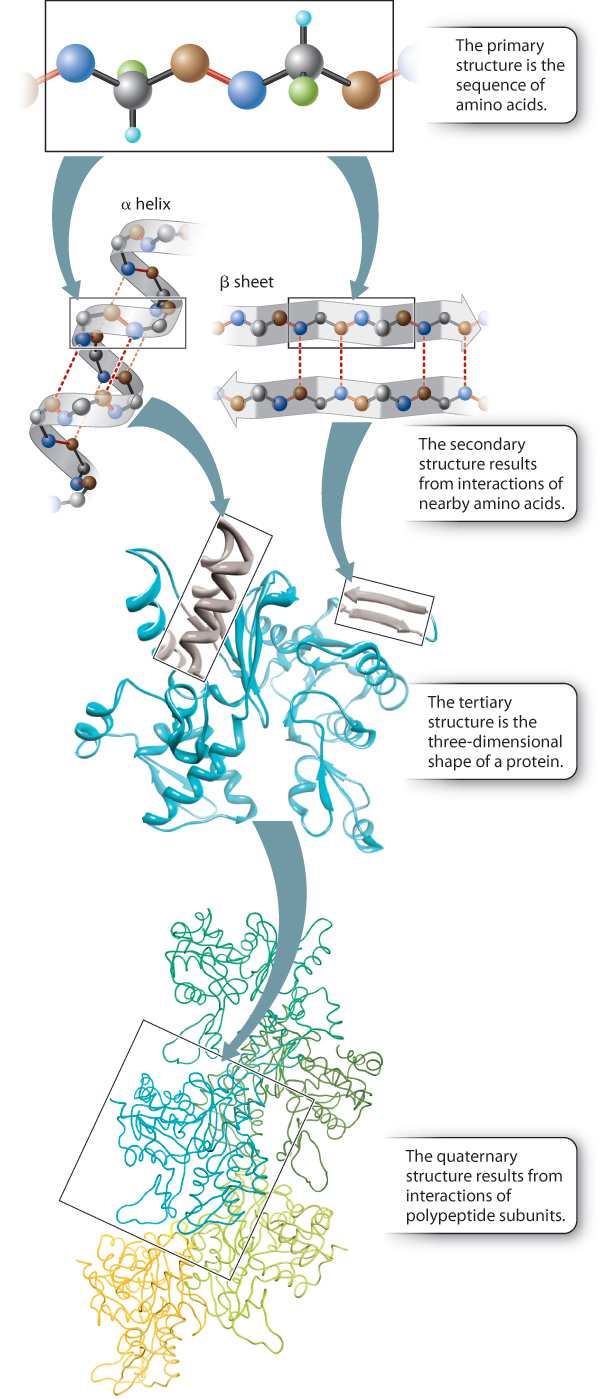
Up to this point, we have considered the sequence of amino acids that make up a protein. This is the first of several levels of protein structure, illustrated in Figure 4.4. The sequence of amino acids in a protein is its primary structure. The sequence of amino acids ultimately determines how a protein folds. Interactions between stretches of amino acids in a protein form local secondary structures. Longer-range interactions between these secondary structures in turn support the overall three-dimensional shape of the protein, which is its tertiary structure. Finally, some proteins are made up of several individual polypeptides that interact with each other, and the resulting ensemble is the quaternary structure.
Proteins have a remarkably wide range of functions in the cell, from serving as structural elements to communicating with the external environment to accelerating the rate of chemical reactions. No matter what the function of a protein is, the ability to carry out this function depends on the three-dimensional shape of the protein. When fully folded, some proteins contain pockets with positively or negatively charged side chains at just the right positions to trap small molecules; others have surfaces that can bind another protein or a sequence of nucleotides in DNA or RNA; some form rigid rods for structural support; and still others keep their hydrophobic side chains away from water molecules by inserting into the cell membrane.
The sequence of amino acids in a protein (its primary structure) is usually represented by a series of three-letter or one-letter abbreviations for the amino acids (abbreviations for the 20 amino acids are given in Figure 4.2). By convention, the amino acids in a protein are listed in order from left to right, starting at the amino end and proceeding to the carboxyl end. The amino and the carboxyl ends are different, so the order matters. Just as TIPS is not the same word as SPIT, the sequence Thr–Ile–Pro–Ser is not the same peptide as Ser–Pro–Ile–Thr.
Quick Check 1
A mutation leads to a change in one amino acid in a protein. The result is that the protein no longer functions properly. How is this possible?
4.1.4 Secondary structures result from hydrogen bonding in the polypeptide backbone.
Hydrogen bonds can form between the carbonyl group in one peptide bond and the amide group in another, thus allowing localized regions of the polypeptide chain to fold. This localized folding is a major contributor to the secondary structure of the protein. In the early 1950s, American structural biologists Linus Pauling and Robert Corey used a technique known as X-ray crystallography to study the structure of proteins. This technique was pioneered by British biochemists Dorothy Crowfoot Hodgkin, Max Petrutz, and John Kendrew, among others (Figure 4.5). Pauling and Corey studied crystals of highly purified proteins and discovered that two types of secondary structure are found in many different proteins. These are the α helix and the β sheet. Both these secondary structures are stabilized by hydrogen bonding along the peptide backbone.
FIG. 4.5What are the shapes of proteins?
BACKGROUND The three-dimensional shapes of proteins can be determined by X-ray crystallography. One of the pioneers in this field was Dorothy Crowfoot Hodgkin, who used this technique to define the structures of cholesterol, vitamin B12, penicillin, and insulin. She was awarded the Nobel Prize in Chemistry in 1964 for her early work. Max Perutz and John Kendrew shared the Nobel Prize in Chemistry in 1962 for defining the structures of myoglobin and hemoglobin using this method.
METHOD X-ray crystallography can be used to determine the shape of proteins, as well as other types of molecules. The first step, which can be challenging, is to make a crystal of the protein. Then X-rays are aimed at the crystal while it is rotated. Some X-rays pass through the crystal, while others are scattered in different directions. A film or other detector records the pattern as a series of spots, which is known as a diffraction pattern. The locations and intensities of these spots can be used to infer the position and arrangement of the atoms in the molecule.
RESULTS The X-ray diffraction pattern for hemoglobin looks like this:

FOLLOW-UP WORK Linus Pauling and Robert Corey used X-ray crystallography to determine two types of secondary structures commonly found in proteins—the α helix and the β sheet. Today, this technique is a common method for determining the shape of proteins.
SOURCES Crowfoot, D. 1935. “X-Ray Single Crystal Photographs of Insulin.” Nature 135:591–592. Kendrew, J.C., G. Bodo, H. M. Dintzis, et al. 1958. “A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray analysis.” Nature 181:662–666. [Photo: Courtesy of William E. Royer, University of Massachusetts Medical School and Vukica Srajer, BioCARS, Center for Advanced Radiation Sources, The University of Chicago]
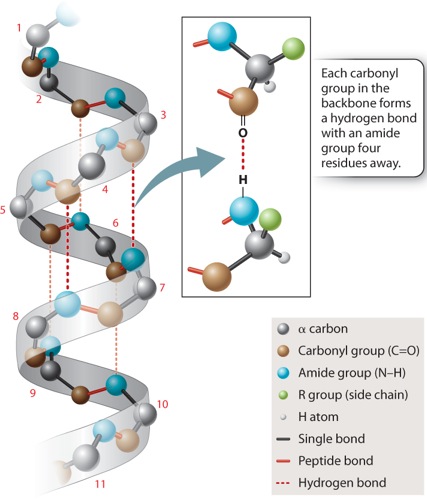
In the α helix shown in Figure 4.6, the polypeptide backbone is twisted tightly in a right-handed coil with 3.6 amino acids per complete turn. The helix is stabilized by hydrogen bonds that form between each amino acid’s carbonyl group and the amide group four residues ahead in the sequence, as indicated by the dashed lines in Figure 4.6. Note that the side chains project outward from the α helix. The chemical properties of the projecting side chains largely determine where the α helix is positioned in the folded protein, and how it might interact with other molecules.
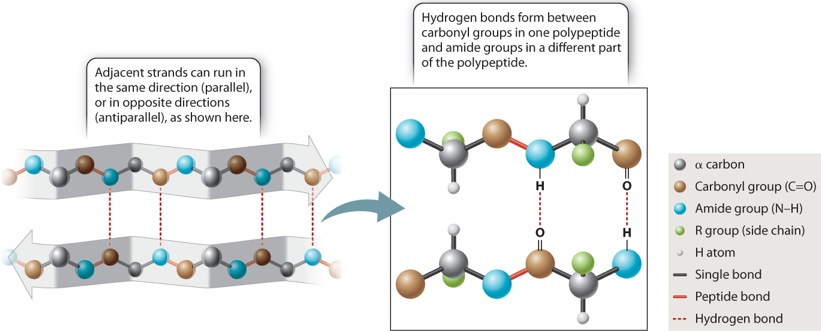
The other secondary structure that Pauling and Corey found is the β sheet, depicted in Figure 4.7. In a β sheet, the polypeptide folds back and forth on itself, forming a pleated sheet that is stabilized by hydrogen bonds between carbonyl groups in one chain and amide groups in the other chain across the way (dashed lines). The side chains (shown in green) project alternately above and below the plane of the β sheet. β sheets typically consist of 4 to 10 polypeptide chains aligned side by side, with the amides in each chain hydrogen-bonded to the carbonyls on either side (except for those at the edges).
β sheets are typically denoted by broad arrows, where the direction of the arrow runs from the amino end of the polypeptide segment to the carboxyl end. In Figure 4.7, the arrows run in opposite directions, and the polypeptide chains are said to be antiparallel. β sheets can also be formed by hydrogen bonding between polypeptide chains that are parallel (pointing in the same direction). However, the antiparallel configuration is more stable because the carbonyl and amide groups are more favorably aligned for hydrogen bonding.
4.1.5 Tertiary structures result from interactions between amino acid side chains.
The tertiary structure of a protein is the three-dimensional conformation of a single polypeptide chain, usually made up of several secondary structure elements. The shape of a protein is defined largely by interactions between the amino acid side chains. By contrast, the formation of secondary structures discussed earlier relies on interactions in the polypeptide backbone and is relatively independent of the side chains. Tertiary structure is determined by the spatial distribution of hydrophilic and hydrophobic side chains along the molecule, as well as by different types of chemical bonds and interactions (ionic, hydrogen, and van der Waals) that form between various side chains. The amino acids whose side chains form bonds with each other may be far apart in the polypeptide chain, but can end up near each other in the folded protein. Hence, the tertiary structure usually includes loops or turns in the backbone that allow these side chains to sit near each other in space and for bonds to form.
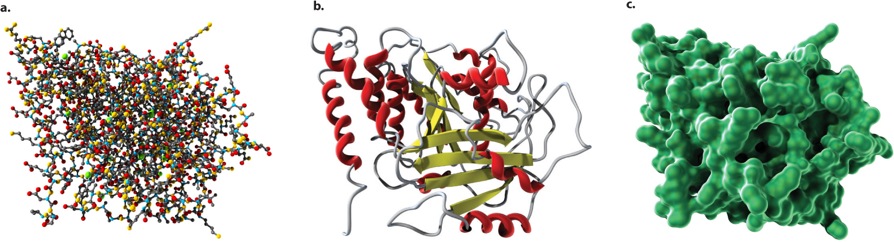
The three-dimensional shapes of proteins can be illustrated in different ways, as shown in Figure 4.8. A ball-and-stick model (Figure 4.8a) draws attention to the atoms in the amino acid chain. A ribbon model (Figure 4.8b) emphasizes secondary structures, with α helices depicted as twisted ribbons and β sheets as broad arrows. Finally, a space-filling model (Figure 4.8c) shows the overall shape and contour of the folded protein.
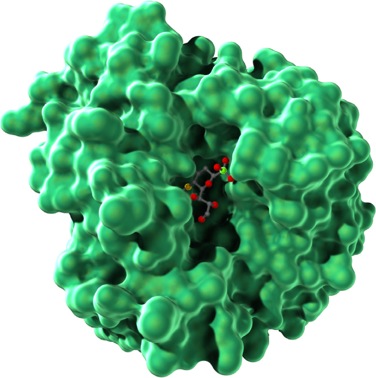
Remember that the folding of a polypeptide chain is determined by the sequence of amino acids. The primary structure determines the secondary and tertiary structures. Furthermore, tertiary structure determines function because it is the three-dimensional shape of the molecule—the contours and distribution of charges on the outside of the molecule and the presence of pockets that might bind with smaller molecules on the inside—that enables the protein to serve as structural support, membrane channel, enzyme, or signaling molecule. Figure 4.9 shows the tertiary structure of a bacterial protein that contains a pocket in the center in which certain side groups can form hydrogen bonds with a specific small molecule and hold it in place.
The principle that structure determines function can be demonstrated by many observations. For example, most proteins can be unfolded, or denatured, by chemical treatment or high temperature, and under these conditions they lose their functional activity; but when the chemicals are removed or the temperature reduced, the proteins refold and their functional activity returns. Similarly, mutant proteins containing an amino acid that prevents proper folding are often devoid of their functional activity.
4.1.6 Polypeptide subunits can come together to form quaternary structures.
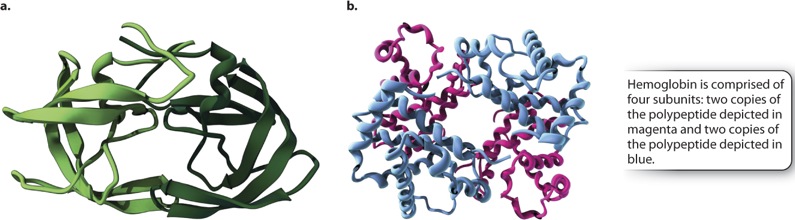
Although many proteins are complete and fully functional as a single polypeptide chain with a tertiary structure, there are many other proteins that are composed of two or more polypeptide chains or subunits, each of which has a tertiary structure, and that also come together to form a higher-order quaternary structure. In the case of a multi-subunit protein, the activity of the complex depends on the quaternary structure formed by the combination of the various tertiary structures.
The polypeptide subunits may be identical or different (Figure 4.10). Figure 4.10a shows an example of a protein produced by HIV that consists of two identical polypeptide subunits. By contrast, many proteins, such as hemoglobin shown in Fig. 410b, are composed of different subunits. In either case, the subunits can influence each other in subtle ways and influence their function. For example, the hemoglobin in red blood cells that carries oxygen has four subunits. When one of these binds oxygen, a slight change in its structure is transmitted to the other subunits, making it easier for them to take up oxygen. In this way, oxygen transport from the lungs to the tissues is improved.
4.1.7 Chaperones help some proteins fold properly.
The amino acid sequence (primary structure) of a protein determines how it forms its secondary, tertiary, and quaternary structures. For about 75% of proteins, the folding process takes place within milliseconds as the molecule is synthesized. Some proteins fold more slowly, however, and for these molecules folding is a dangerous business. The longer these polypeptides remain in a denatured state, the longer their hydrophobic groups are exposed to other macromolecules in the crowded cytoplasm. The hydrophobic effect, along with van der Waals interactions, tends to bring the exposed hydrophobic groups together, and their inappropriate aggregation may prevent proper folding. Correctly folded proteins can sometimes unfold because of elevated temperature, for example, and in the denatured state they are subject to the same risks of aggregation.
Cells have evolved proteins called chaperones that help protect slow-folding or denatured proteins until they can attain their proper three-dimensional structure. Chaperones bind with hydrophobic groups and nonpolar side chains to shield them from inappropriate aggregation, and in repeated cycles of binding and release they give the polypeptide time to find its correct shape.