13.1 GENOME SEQUENCING
What exactly is a genome? Originally, the term referred to the complete set of chromosomes present in a reproductive cell, like a sperm or an egg, which in the human genome is 23 chromosomes. The word “genome” is almost as old as the word “gene,” and it was coined at a time when chromosomes were thought to consist of densely packed genes lined up one after another.
We know now, however, that chromosomes consist primarily of DNA and associated proteins, and that the genetic information in the chromosomes resides in the DNA. One might therefore define the genome as the DNA molecules that are transmitted from parents to offspring. This definition has the advantage of including the DNA in organisms that lack true chromosomes, such as bacteria and archaeons, as well as eukaryotic organelles that contain their own DNA, such as mitochondria and chloroplasts. But a definition restricted to DNA is too narrow because it excludes viruses, like HIV, whose genetic material consists of RNA. Defining a genome as the genetic material transmitted from parents to offspring therefore embraces all known cellular forms of life, all known organelles, and all viruses.
In this first section, we focus on how genomes are sequenced, building on the DNA sequencing technology introduced in Chapter 12.
13.1.1 Complete genome sequences are assembled from smaller pieces.
In Chapter 12, we discussed a method of DNA sequencing known as Sanger sequencing. Such methods are now automated to the point where specialized machines can determine the sequence of billions of DNA nucleotides in a single day. However, even with recent advances, the data are obtained in the form of short sequences, typically less than a few hundred nucleotides long. If you are interested in sequencing a short DNA fragment, these technologies work well. However, let’s say you are interested in the sequence of human chromosome 1, which is a DNA molecule approximately 250 million nucleotides long. How can you sequence a DNA molecule as long as that?
In one approach, the single long DNA molecule is first broken up into small fragments, each of which is short enough to be sequenced by existing technologies. Even though the sequence data obtained from a small DNA fragment is only a minuscule fraction of the length of the DNA molecules in most genomes, each run of an automated sequencing machine yields hundreds of millions of these short sequences from random locations throughout the genome. To sequence a whole genome, researchers typically sequence such a large number of random DNA fragments that, on average, any particular small region of the genome is sequenced 10–50 times. This redundancy is necessary to minimize the number of errors present in the final genome sequence and to minimize the number and size of gaps where the genome sequence is incomplete.
When a sufficient number of short sequences has been obtained, the next step is sequence assembly: The short sequences are put together in the correct order to generate the long, continuous sequence of nucleotides in the DNA molecule present in each chromosome.
Assembly is accomplished by complex computer programs, but the principle is simple. The short sequences are assembled according to their overlaps, as illustrated in Fig. 13.1, using a sentence to represent the nucleotide sequence. When assembly is complete, the overlapping fragments yield the famous sentence from Watson and Crick’s original paper on the chemical structure of DNA, in which, referring to the pairing of bases A with T and of G with C, they write, “It has not escaped our notice that the specific pairing we have postulated immediately suggests a plausible copying mechanism for the genetic material.” This approach is called shotgun sequencing because the sequenced fragments do not originate from a particular gene or region but from sites scattered randomly across the chromosome.
FIG. 13.1How are whole genomes sequenced?
BACKGROUND DNA sequencing technologies can only determine the sequence of DNA fragments far smaller than the genome itself. How can the sequences of these small fragments be used to determine the sequence of an entire genome? In the early years of genome sequencing, many researchers thought that it would be necessary to know first where in the genome each fragment originated before sequencing it. A group at Celera Genomics reasoned that, if so many fragments were sequenced that the ends of one would almost always overlap with those of others, then a computer program with sufficient power might be able to assemble the short sequences to reveal the sequence of the entire genome.
HYPOTHESIS A genome sequence can be determined by sequencing small, randomly generated DNA fragments and assembling them into a complete sequence by matching regions of overlap between the fragments.
EXPERIMENT Hundreds of millions of short sequences from the genome of the fruit fly, Drosophila melanogaster, were sequenced. Fig. 13.1a shows examples of overlapping fragments, using an English sentence as an analogy.
RESULTS The computer program the group had written to assemble the fragments worked. The researchers were able to sequence the entire Drosophila genome by piecing together the fragments according to their overlaps. In the sentence analogy, the fragments (Fig. 13.1a) can be assembled into the complete sentence (Fig. 13.1b) by matching the overlaps between the fragments.

CONCLUSION The hypothesis was supported: Celera Genomics could determine the entire genomic sequence of an organism by sequencing small, random fragments and piecing them together at their overlapping ends.
FOLLOW-UP WORK Today, the computer assembly method is routinely used to determine genome sequences. This method is also used to infer the genome sequences of hundreds of bacterial species simultaneously—for example, in bacterial communities sampled from seawater or from the human gut.
SOURCE Adams, M. D., et al. 2000. “The Genome Sequence of Drosophila melanogaster.” Science 287:2185–2195.
Quick Check 1
DNA sequencing technology has been around since the late 1970s. Why did sequencing whole genomes present a challenge?
13.1.2 Sequences that are repeated complicate sequence assembly.
Sequence assembly is not quite as straightforward as Fig. 13.1 might suggest. Real sequences are composed of nucleotides with only the four bases designated by the four letters A, T, G, and C, and any given short sequence could come from either strand of the double-stranded DNA molecule. Therefore, the overlaps between fragments must be long enough both to ensure that the assembly is correct, and to determine from which strand of DNA the short sequence originated.
Some features of genomes present additional challenges to sequence assembly, and the limitations of the computer programs for handling such features require hands-on assembly. Chief among these complicating features is the problem of repeated sequences.
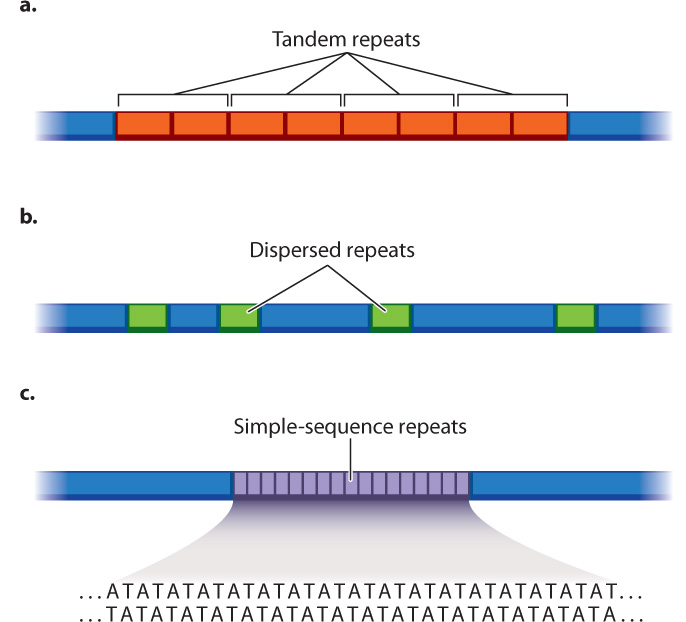
There are a variety of types of repeated sequence in eukaryotic genomes, and some are shown in Fig. 13.2. The repeated sequence may be several thousand nucleotides long and present in multiple identical or nearly identical copies. These long repeated sequences may be tandem, meaning that they are next to each other (Fig. 13.2a), or they may be dispersed throughout the genome (Fig. 13.2b).
The difficulty with long repeated sequences is that they typically are much longer than the short sequences obtained by automated sequencing. As a result, the repeat may not be detected at all. And if the repeat is detected, there is no easy way of knowing the number of copies of the repeat, that is, whether the segment includes two, three, four, or any number of copies of the repeat. Sometimes, researchers can use the ends of repeats, where the fragments overlap with an adjacent, nonrepeating sequence, as a guide to the position and number of repeats.
In another type of repeat, the repeating sequence is short, even as short as two nucleotides, such as AT, repeated over and over again in a stretch of DNA (Fig. 13.2c). Short repeating sequences of this kind are troublesome for sequencing machines because any single-stranded fragment consisting of alternating AT can fold back upon itself to form a double-stranded structure in which A is paired with T. Such structures are more stable than the unfolded single-stranded structures and are not easily sequenced.
Quick Check 2
Let’s say that a stretch of repeated AT is successfully sequenced. From what you know of the difficulties of sequencing long repeated sequences, what other problems might you encounter in assembling these fragments?
Case 3 You, From A To T: Your Personal Genome
13.1.3 Why sequence your personal genome?
The Human Genome Project, which began in 1990, had the goal of sequencing the human genome as well as the genomes of certain key organisms used as models in genetic research. The model organisms chosen are the mainstays of laboratory biology—a species each of bacteria, yeast, nematode worm, fruit fly, and mouse. By 2003, the genome sequences of these model organisms had been completed, as well as the human genome. By then, the cost of sequencing had become so low and the sequence output so high that many more genomes were sequenced than originally planned. Large-scale genome sequencing is still done today, and soon you could choose to have your personal genome sequenced.
Why sequence more genomes? And if the human genome is sequenced, why sequence yours? As we saw in Case 3: You, From A to T, there is really no such thing as the human genome, any more than there is the fruit fly genome or the mouse genome. With the exception of identical twins, every person’s genome is unique, the product of a fusion of a unique egg with a unique sperm. The sequence that is called “the human genome” is actually a composite of sequences from different individuals. This sequence is nevertheless useful because most of us share the same genes and regulatory regions, organized the same way on chromosomes.
Detailed knowledge of your own personal genome can be valuable. Our individual DNA sequences differ at millions of nucleotide sites from one person to the next. Some of these differences account in part for the physical differences we see among us; others have the potential to predict susceptibility to disease and response to medication. For Claudia Gilmore, knowledge of the sequence of her BRCA1 gene had a significant impact on her life (Case 3: You, From A to T).
Determining these differences is a step toward personalized medicine, in which an individual’s genome sequence, by revealing his or her disease susceptibilities and drug sensitivities, allows treatments to be tailored to the individual. There may come a time, perhaps within your lifetime, when personal genome sequencing becomes part of routine medical testing. Information about a patient’s genome will bring benefits but also raises ethical concerns and poses risks to confidentiality and insurability.