15.1 GENOTYPE AND PHENOTYPE
We tend to think of mutations as something negative or harmful; the term “mutant” ordinarily connotes something unpleasantly abnormal. But because mutations result in genetic variation among individuals and organisms, we are in fact all mutants, different from one another genetically because of mutations, that is, differences in our DNA. Whereas some mutations are harmful, some have no effect on an organism, and some indeed are beneficial. Without mutations, evolution would not be possible. Mutations generate the occasional favorable variants that allow organisms to evolve and become adapted to their environment over time (Chapter 21).
15.1.1 Genotype is the genetic makeup of a cell or organism; phenotype is its observed characteristics.
The genetic makeup of a cell or organism constitutes its genotype. A population with a gene pool that has many variants in many different genes will consist of organisms with numerous different genotypes. For example, any two human genomes are likely to differ at about 3 million nucleotide sites. Most humans have the same genes as one another, organized in the same way along the same set of chromosomes. We differ primarily from one person to the next in a very small fraction of nucleotides.
Mutations, as we have seen, are the ultimate source of differences among genotypes. If we consider any present-day population of organisms, we find that some mutations are very common. Geneticists use the term polymorphism to refer to any genetic difference among individuals that is sufficiently common that it would almost certainly be present in a group of 50 randomly chosen individuals. For example, if many people have an A–T pair at a particular site in the genome, but many others have a G–C pair at the same site, this difference is a polymorphism. Of course, the polymorphism is the result of a mutation. All individuals once had the same genotype at this site, but a mutation in one or a few individuals made different genotypes possible. In this example, the mutation that made either A–T or G–C possible occurred sometime in the past, and is now commonly found in the population.
Phenotype is an individual’s observable characteristics or traits, such as a person’s height, weight, eye color, and so forth. The phenotype may be visible, as in these characteristics, or may be seen in the development, physiology, or behavior of a cell or organism. For example, color blindness and lactose intolerance are phenotypes. The phenotype results in part from the genotype: A genotype with a mutated gene for an enzyme that would normally metabolize lactose can lead to the phenotype of lactose intolerance. However, the environment also commonly plays an important role, so it is most accurate to say that a phenotype results from an interaction between the genotype and environment. These genotype–environment interactions are discussed in Chapter 18.
15.1.2 The effect of a genotype often depends on several factors.
Let’s examine how genotype influences phenotype with an example of a genetic polymorphism that we introduced in the previous chapter. Fig. 15.1 shows genetic variation in the gene for β-(beta-) globin, one of the subunits of hemoglobin that carries oxygen in red blood cells. Three forms of the β-globin gene are shown in the figure: A, S, and C. These three forms of the gene are relatively common in certain African populations and in people of African descent (the mutations originated in Africa). The different forms of any gene are called alleles, and they correspond to different DNA sequences (polymorphisms) in the genes. In this case, the most common allele is the A allele, which has a GAG codon in the position indicated. This codon translates to glutamic acid in the resulting polypeptide.
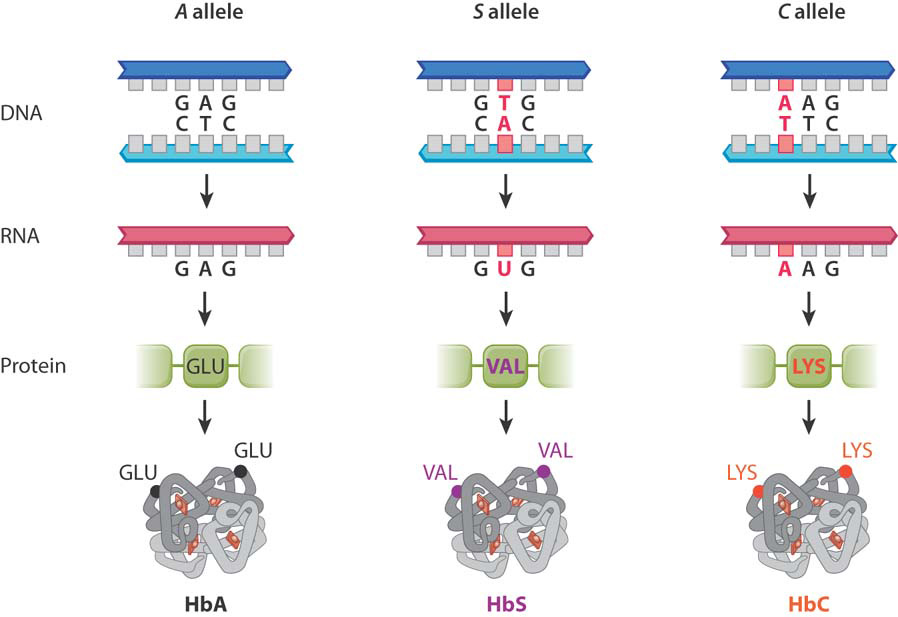
The allele denoted S in Fig. 15.1 is associated with sickle-cell anemia (Chapter 14). In this allele, the GAG codon seen in the A allele is instead a GTG codon, with the result that the glutamic acid in the protein is replaced with valine (Val). The third allele in Fig. 15.1 is the C allele. It has a variation in exactly the same codon as the S allele, but in this case the change is from the normal GAG to AAG, which results in glutamic acid being replaced with lysine (Lys). Although in each case only one amino acid of the β-globin protein is affected, this change can have a dramatic effect on the function of the protein, since the amino acid sequence determines how a protein folds, and protein folding in turn determines the protein’s function (Chapter 4).
An individual who inherits an allele of the same type from each parent is said to be homozygous. For the hemoglobin A, S, and C alleles, there are three possible homozygous genotypes, AA, SS, and CC. (The first letter or symbol indicates the allele inherited from one parent, and the second indicates the allele inherited from the other parent. By convention, alleles and genotypes are designated by italic letters.)
By contrast, individuals who inherit different types of alleles from their parents are heterozygous. For the hemoglobin A, S, and C alleles, there are three possible heterozygous genotypes: AS, AC, and SC. Note that while each individual can have only two alleles of a gene, many more alleles can exist in an entire population. In the hemoglobin example, a person can have two identical alleles (homozygous) or two different alleles (heterozygous), but there are three different alleles available in the population to mix and match in each individual. How those alleles are inherited is explored in Chapter 16.
What are the effects of these mutations? Are they beneficial, harmful, or neutral? The short answer is, it depends. Let’s consider the S allele. When it is inherited from both parents, the individual is homozygous SS and has sickle-cell anemia. In the absence of proper medical care, patients with sickle-cell anemia usually die before adulthood. However, when the S allele is inherited from one parent and the A allele is inherited from the other parent, the individual is heterozygous (AS) and the mutant allele causes only a mild form of blood disease. Furthermore, in Africa, where malaria is widespread, the mutation is actually beneficial because it affords partial protection against severe malaria.
Whether the effects of an allele, in this case the S allele, are beneficial or harmful illustrates two important principles about the connection between genotype and phenotype. First, the answer often depends on whether the mutation is homozygous or heterozygous. In areas with malaria, the S allele is harmful as a homozygote but beneficial as a heterozygote. Second, the effect of a particular genotype may depend on the environment. The S allele, as a heterozygote, is beneficial only in malarial-prone regions, where it offers protection from the disease that outweighs its other effects. In areas without malaria, it is harmful.
What about the C allele? Individuals who inherit a C allele from one parent and an A allele from the other parent (genotype AC) are partially protected against severe malaria, and those who inherit a C allele from both parents (genotype CC) are not only more protected from malaria but also have at worst a mild anemia that usually needs no medical treatment. As with the S allele, the phenotype of an allele may depend on whether the allele was inherited from one or both parents.
Quick Check 1
A mutation arises in a bacterium that confers antibiotic resistance. Is this mutation harmful, beneficial, or neutral?
15.1.3 Some genetic differences are major risk factors for disease.
As we saw in the last chapter, when a mutation occurs in the coding sequence of a gene, it may have no effect on the amino acid sequence, or it may result in a change in the amino acid sequence, introduce a stop codon, or shift the reading frame. Many of these latter mutations are harmful. Harmful mutations are often eliminated in one or a few generations because they decrease the survival and reproduction of the individuals that carry them.
Sometimes, however, harmful mutations persist in a population. For example, many polymorphisms increase susceptibility to particular diseases. One such polymorphism increases the risk of emphysema, a condition marked by overinflation of the air sacs in the lungs. Emphysema patients suffer from shortness of breath even when at rest, a wheezy cough, and increased blood pressure in the arteries of the lungs. Without proper treatment, the disease progresses to respiratory failure or congestive heart failure, and eventual death. Onset of the disease usually occurs in middle age, and the lifespan of affected individuals is shortened by 10 to 30 years depending on the effect of treatment.
About 80% of all cases of emphysema are associated with cigarette smoking, which affects the action of the enzyme alpha-1 antitrypsin (α1AT). The main function of α1AT is to inhibit another enzyme, known as elastase (Fig. 15.2a). The elasticity of the lung, which allows normal breathing, requires a balance between the production and the breakdown of the connective-tissue protein elastin. Breakdown of elastin results from the action of elastase. Too-rapid breakdown is normally prevented by the inhibition of elastase by α1AT, but cigarette smoke reduces the activity of α1AT (Fig. 15.2b). The result is excessive destruction of elastin, loss of lung elasticity, and emphysema.
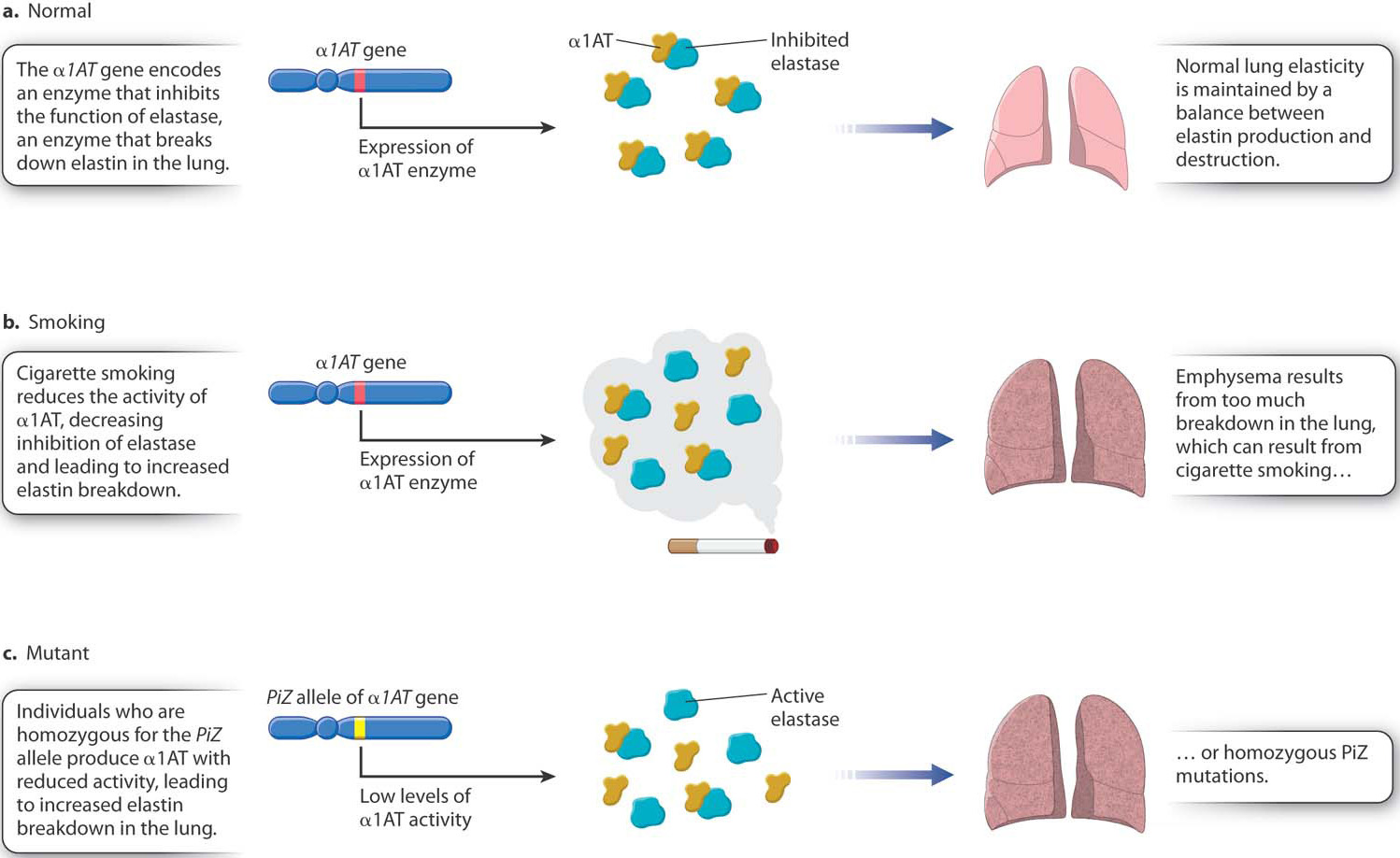
While most cases of emphysema are due to smoking, not everyone who smokes will get emphysema. A smoker’s chance of getting emphysema is significantly increased by inheriting a mutation in the gene that encodes α1AT. Among the many different alleles of this gene, a defective allele denoted PiZ is particularly common in populations of European descent. Among Caucasians in the United States, about 1 person in 30 is heterozygous for the PiZ allele, and about 1 in 3000 is homozygous PiZ/PiZ. Individuals with the homozygous genotype produce α1AT with reduced activity and hence have reduced elastase inhibition, leading to severe emphysema and death in more than 70% of affected individuals (Fig. 15.2c). Smoking markedly increases both the severity of the disease and the rapidity of its progression in PiZ/PiZ individuals. The life expectancy of PiZ/PiZ nonsmokers is 65 years, whereas that of PiZ/PiZ smokers is only 40 years.
This is an example of genotype-by-environment interaction, in which a phenotype is the result of an interplay between genes and the environment. In this case, a particular combination of genetic and environmental risk factors is much worse than either risk factor acting alone. Chapter 18 includes a detailed discussion of genotype-by-environment interaction.
15.1.4 Not all genetic differences are harmful.
Some mutations have no effect on the organism, or have effects that are not associated with differences in survival or reproduction. Such mutations are considered neutral. Neutral mutations are often found in noncoding DNA, and so occur especially in organisms with large genomes and abundant noncoding DNA (Chapter 13).
Quick Check 2
Given what you read about the human genome in Chapter 13, would you predict that most mutations in humans are harmful, beneficial, or neutral?
Sometimes common, harmless genetic variations occur in coding sequences. One example in human populations is the taster phenotype associated with perception of a bitter taste from certain chemicals, including phenylthiocarbamide (PTC). The taster polymorphism was discovered in 1931 when a commercial chemist seeking a new artificial sweetener accidentally released a cloud of fine crystalline PTC and heard his colleague working nearby complain about its bitter taste. The chemist himself tasted nothing. He began testing his own and other families, and set the stage for future genetic studies.
The minimal concentration for PTC tasting varies almost continuously among individuals, but being able to taste a concentration of 0.5 millimolar or less is often taken as the cutoff between the taster and nontaster phentoypes. In a sample from Utah, the frequency of nontasters is about 30%. This is typical for people of European descent, but the frequency of nontasters differs among populations, from as low as 3% in West Africa to as high as 40% in India.
The ability or inability to taste PTC is due largely, but not exclusively, to alleles of a single gene that encodes a taste receptor in the tongue. Nonhuman primates are homozygous for an allele of this gene known as the PAV allele, so called because the protein it encodes has the amino acids proline (P), alanine (A), and valine (V) at specific positions. Humans also have the PAV allele, and PAV/PAV homozygous genotypes are almost all tasters.
The most common allele associated with the nontaster phenotype is AVI, in which the amino acids in the taste receptor are alanine (A), valine (V), and isoleucine (I) instead of proline, alanine, and valine. About 80% of homozygous AVI/AVI genotypes are nontasters. One copy of the PAV allele is usually sufficient for the taster phenotype; about 98% of PAV/AVI heterozygous genotypes are tasters.
The factors contributing to the taster phenotype are more complex than a genotype at a single gene, however. The AVI/AVI genotype tips the balance toward the nontaster phenotype, but not completely. Other genes and the environment also play a role.
Why is this strange variation present in the human population? One hypothesis is that an aversion to compounds that contain a thiourea group (N–C=S), as PTC does, may discourage eating certain plants that produce poisonous defense compounds. One class of such compounds is the glycosinolates, which are present in many wild plants and some cultivated vegetables, including broccoli, watercress, turnip, and horseradish. Sure enough, PAV/PAV tasters rate such vegetables as significantly more bitter than AVI/AVI nontasters, whereas PAV/AVI heterozygous genotypes are intermediate in their perception of bitterness.
We hasten to add, however, that the low level of glycosinolates in broccoli and other cultivated vegetables is nontoxic, so you should still eat your vegetables! But if you find broccoli and its relatives somewhat bitter, you may well be a taster.
15.1.5 A few genetic differences are beneficial.
While many mutations are neutral, or nearly so, and many others are harmful, some mutations are beneficial. In human populations, beneficial mutations are often discovered through their effects in protecting against infectious disease. The most widely known example is probably the sickle-cell allele in the gene encoding the β chain of hemoglobin, which when heterozygous protects against malaria (see Fig. 15.1). In this section, we consider another example that protects against AIDS, which is caused by the human immunodeficiency virus (HIV).
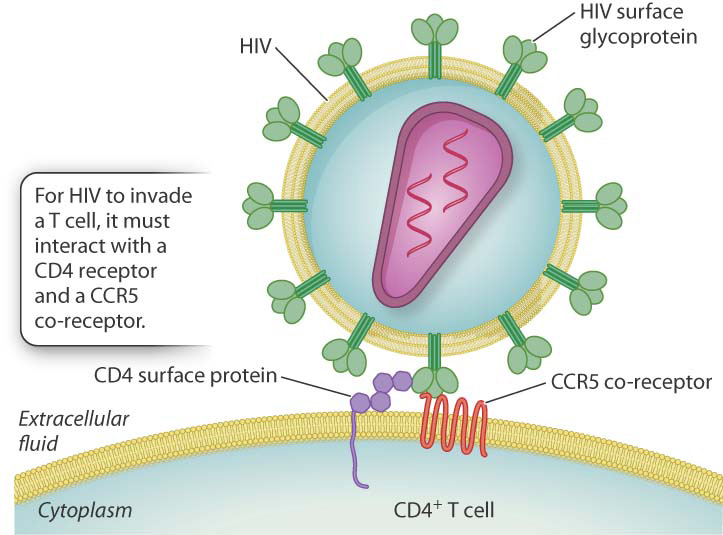
By means of its surface glycoprotein (a product of the env region in the annotated HIV genome shown in Fig. 13.7), HIV combines with a cell-surface receptor called CD4 to gain entry into T cells. Interaction with CD4 alone, however, does not enable the virus to infect the T cell. The surface glycoprotein must also interact with another receptor, which is denoted CCR5, in the early stages of infection (Fig. 15.3). The normal function of CCR5 is to bind certain small secreted proteins that promote tissue inflammation in response to infection. But because CCR5 is also an HIV receptor, cells lacking CCR5 are more difficult to invade.
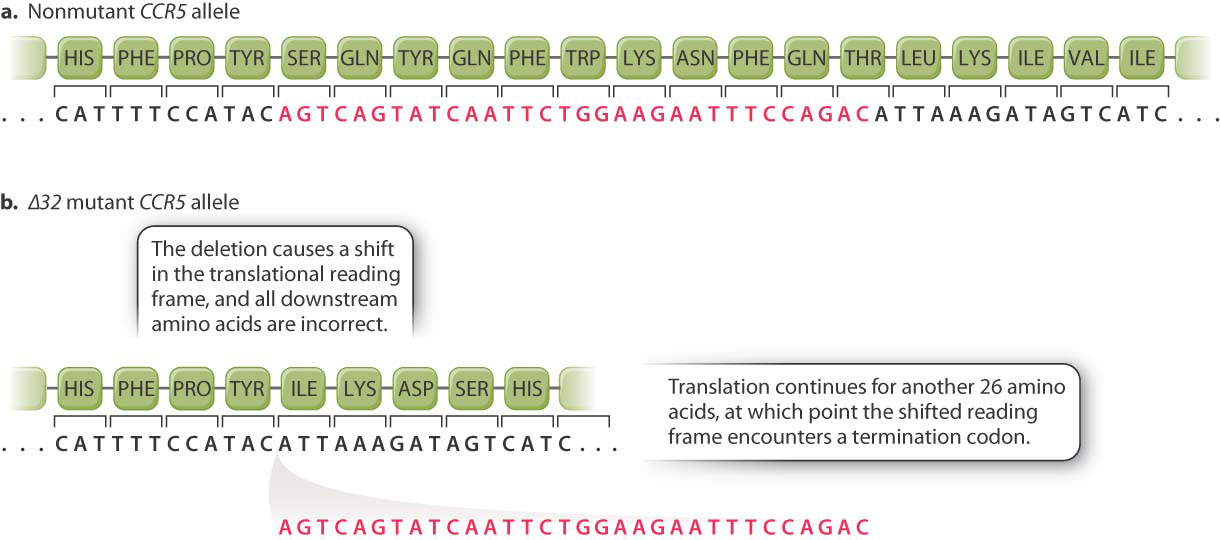
A beneficial effect of a particular mutation in the CCR5 gene was discovered in studies focusing on HIV patients whose infection had not progressed to full-blown AIDS for 10 years or more. The protective allele is denoted the Δ32 allele because the mutation is a 32-base-pair deletion in the coding sequence of the CCR5 gene (Fig. 15.4). Because the 32 is not a multiple of 3, the reading frame for translation is shifted at the site of the deletion, and instead of the normal amino acid sequence Ser–Gln–Tyr–Gln–Phe ⋯, the mutant sequence is Ile–Lys–Asp–Ser–His ⋯. Not only is the amino acid sequence incorrect, the ribosome encounters a stop codon a mere 26 amino acids farther along and translation terminates. The mutant protein is 215, not 352, amino acids long. The CCR5 protein produced by the Δ32 allele is completely inactive.
The effect of the Δ32 allele is pronounced. In homozygous Δ32/Δ32 genotypes, HIV progression to AIDS is rarely observed. There is some protection even in heterozygous Δ32 genotypes, where progression to AIDS is delayed by an average of about 2 years.
Much has been written about the evolutionary history of the Δ32 allele. It is found almost exclusively in European populations, where the frequency of heterozygous genotypes ranges from 10% to 25%. The narrow geographical distribution was originally interpreted to mean that the allele was selected over time because it provided protection against some other infectious agent that also interacted with the CCR5 protein. Both the bacterium that caused the Black Death, which roared through Europe beginning in about 1350, and smallpox virus have been suggested.
Beneficial mutations not only provide protection against disease; in rare cases, they permit organisms to become adapted to their environment. For example, certain birds, such as Rüppell’s vulture, commonly fly at altitudes of 20,000 feet and sometimes much higher. This feat is possible because of mutations in the structure of hemoglobin that allow hemoglobin to bind oxygen with high affinity, even at the low pressure of oxygen high in the atmosphere. These mutations were selected and passed on generation after generation, allowing the birds to be well adapted to flying at high altitudes.