15.4 GENETIC VARIATION IN CHROMOSOMES
Copy-number variation usually involves only one or a small number of genes, and the size of the duplicated or deleted region is physically so small that the differences are undetectable with conventional microscopy. In the human genome, some common variants involve large regions of the chromosome and are big enough to be visible through a microscope. Most of these variations involve regions of the genome with an extremely low density of genes, such as regions around the centromere or the long arm of the Y chromosome.
It may come as a surprise to learn that major chromosomal differences occur quite commonly. They are infrequently observed, however, because most of them are lethal. Nevertheless, a few major chromosomal differences are found in the general population. Some are common enough that their phenotypic effects are familiar. Others are less common, and a few have no major phenotypic effects at all. In this section, we focus on the types of difference that are observed, what causes them, and their phenotypic effects.
15.4.1 Nondisjunction in meiosis results in extra or missing chromosomes.
Nondisjunction is the failure of a pair of chromosomes to separate during anaphase of cell division (Chapter 11). The result is that one daughter cell receives an extra copy of one chromosome, and the other daughter cell receives no copy of that chromosome. Nondisjunction can take place in mitosis, and it leads to cell lineages with extra or missing chromosomes—which is often observed in cancer cells.
When nondisjunction takes place in meiosis, the result is reproductive cells (gametes) that have either an extra or a missing chromosome. Fig. 15.12 illustrates nondisjunction in meiosis. Fig. 15.12a shows key stages in a normal meiosis, in which chromosome separation (disjunction) takes place in both meiotic divisions, with the result that each gamete receives one and only one copy of each chromosome.
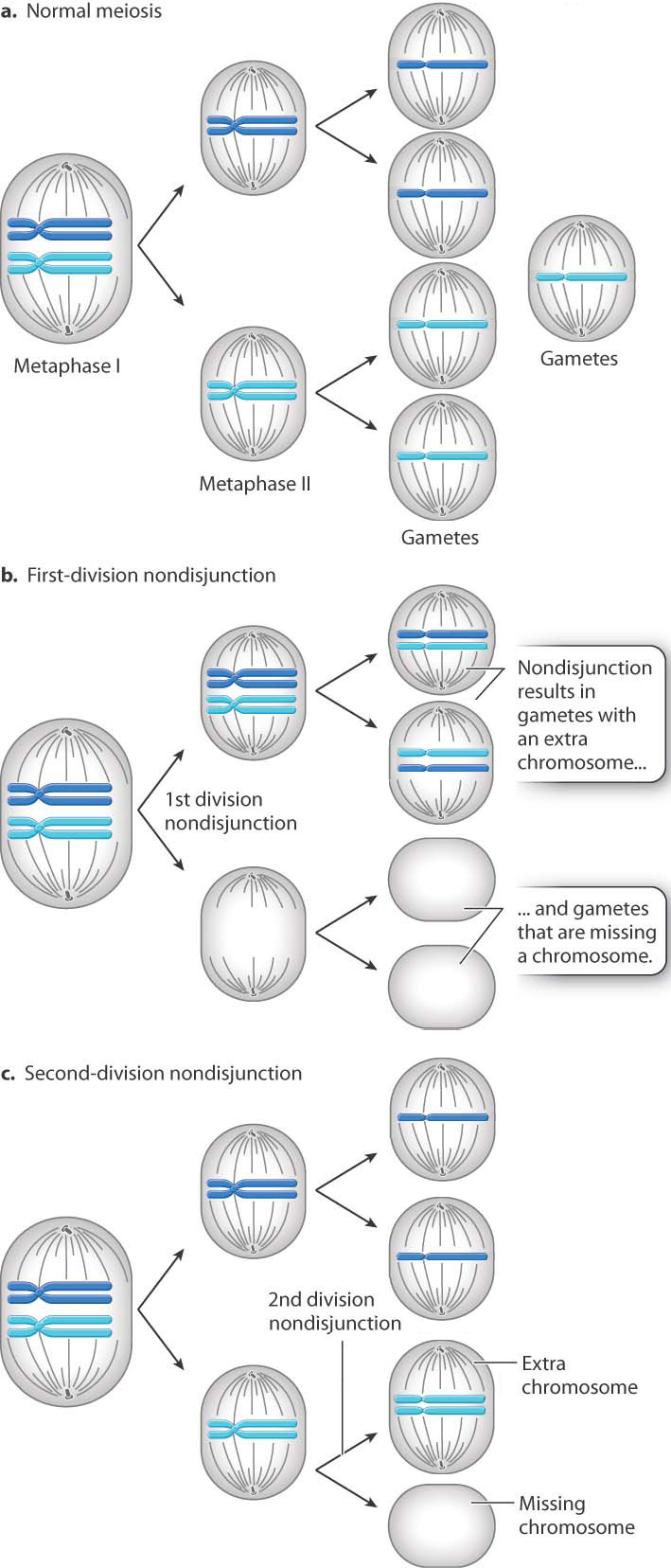
When failure of chromosome separation occurs, it can take place in either of the two meiotic divisions. Both types of nondisjunction result in gametes with an extra chromosome or a missing chromosome, but there is an important difference. In first-division nondisjunction (Fig. 15.12b), the homologous chromosomes fail to separate and so the chromosomes that move together are not genetically identical. On the other hand, in second-division nondisjunction (Fig. 15.12c), the sister chromatids of a chromosome fail to separate and therefore the two copies of the chromosome that end up in the same gamete are genetically identical. (In this discussion, we are ignoring the process of crossing over discussed in Chapter 11.)
15.4.2 Some human disorders result from nondisjunction.
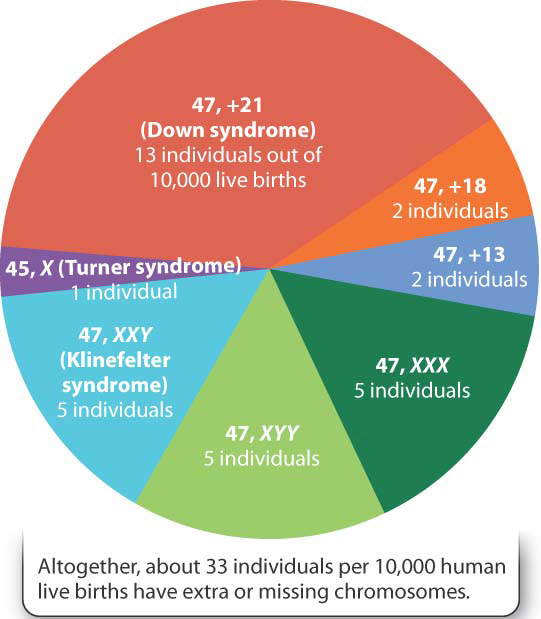
Approximately 1 out of 155 live-born children (0.6%) has a major chromosome abnormality of some kind. About half of these have a major abnormality in chromosome structure, such as an inversion or translocation (Chapter 14). The others have a chromosome that is present in an extra copy or a chromosome that is missing because of nondisjunction—a parent has produced an egg or a sperm with an extra or a missing chromosome (Fig. 15.13).
Perhaps the most familiar of the conditions resulting from nondisjunction is Down syndrome, which results from the presence of an extra copy of chromosome 21. Down syndrome is also known as trisomy 21 because affected individuals have three copies of chromosome 21. The extra chromosome can be seen in a chromosome layout called a karyotype. French researchers Jérôme Lejeune, Marthe Gautier, and Raymond Turpin first saw the extra chromosome when they developed a technique to visualize a patient’s chromosomes (Fig. 15.14). The shorthand for the condition is 47,+21, meaning that the individual has 47 chromosomes with an extra copy of chromosome 21.
FIG. 15.14What is the genetic basis of Down syndrome?
BACKGROUND The detailed images of cells and chromosomes available today are produced by microscopic and imaging techniques not available during most of the history of genetics and cell biology. Techniques for observing human chromosomes were originally so unreliable that the correct number of human chromosomes was firmly established only in the mid-1950s. Mistakes were common, such as counting two nearby chromosomes as one or by including chromosomes in the count of one nucleus when they actually belonged to a nearby nucleus. Despite these limitations, in 1959 French scientists Jérôme Lejeune, Marthe Gautier, and Raymond Turpin decided to investigate whether any common birth abnormalities were associated with chromosomal abnormalities. They chose Down syndrome, one of the most common birth abnormalities, because its genetic basis was an enigma.

HYPOTHESIS Down syndrome is associated with extra or missing chromosomes.
EXPERIMENT The chromosomes of five boys and four girls with Down syndrome were studied. A small patch of skin was taken from each child and placed in growth medium. In this medium, fibroblast cells, which synthesize the extracellular matrix that provides the structural framework of the skin, readily undergo DNA synthesis and divide. After a period allowing cellular growth and division, the fibroblast cells were prepared in a way that allowed the researchers to count the number of chromosomes under a microscope.
RESULTS Chromosomes were counted in 103 cells, yielding counts of 46 chromosomes (10 cells), 47 chromosomes (84 cells), and 48 chromosomes (9 cells). For each cell examined, the researchers noted whether the count was “doubtful” or “absolutely certain.” The researchers marked cells as “doubtful” if the chromosomes or nuclei were not well separated from one another. They encountered 57 cells of which they felt “absolutely certain,” and in each of these they counted 47 chromosomes. The extra chromosome was always one of the smallest chromosomes in the set.
CONCLUSION The researchers concluded that Down syndrome is the result of a chromosomal abnormality, an extra copy of one of the smallest chromosomes. The discovery helped resolve many of the questions surrounding the genetics of Down syndrome.
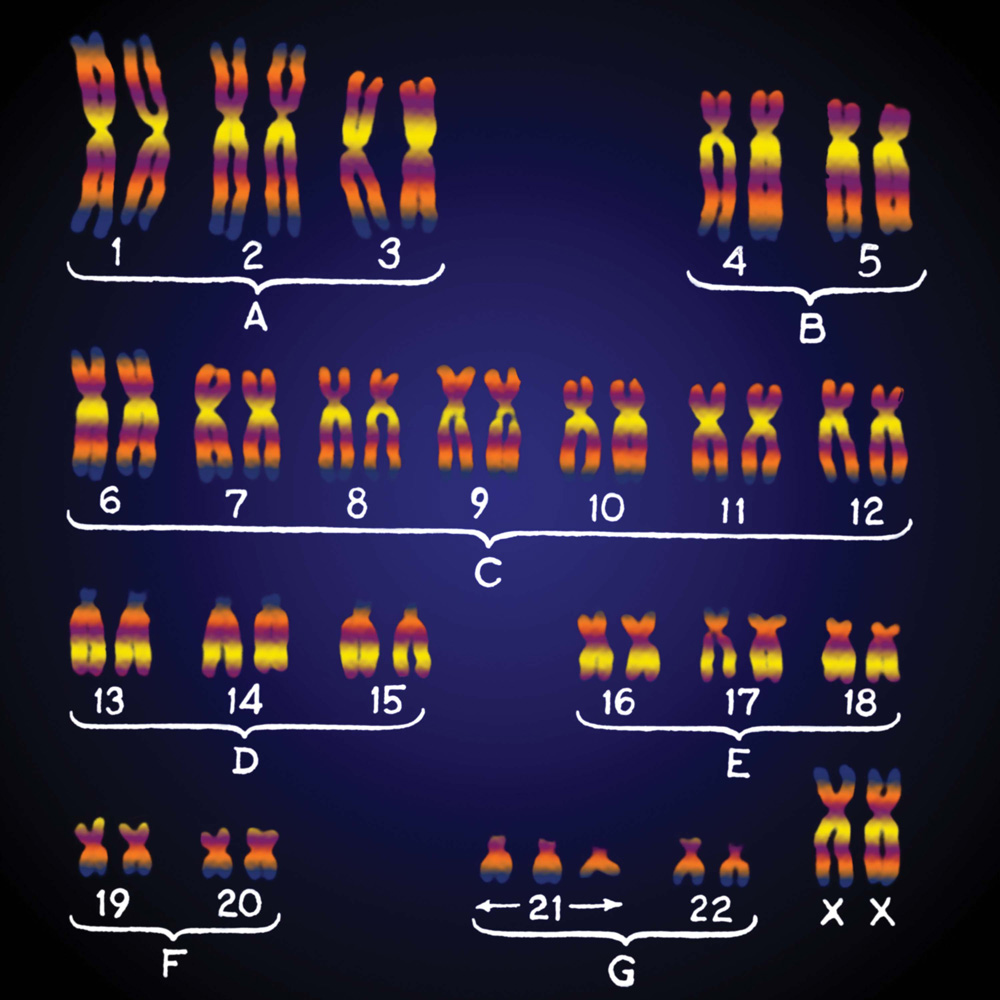
FOLLOW-UP WORK Improved techniques including chromosome banding (shown in Fig. 13.15) later indicated that the extra chromosome was chromosome 21. The finding stimulated many other chromosome studies. Today it is clear that, except for the sex chromosomes, the vast majority of fetuses with extra or missing chromosomes undergo spontaneous abortion. Furthermore, the Human Genome Project has indicated that genes on chromosome 21 have direct roles in many of the signs and symptoms of Down syndrome.
SOURCE J. Lejeune, Gautier, M., and Turpin, R. 1959. “Premier exemple d’aberration autosomique humaine.” Comptes rendus des séances de l’Académie des Sciences. 248:1721–1722. Translated from the French in Landmarks in Medical Genetics, edited by P. S. Harper. Oxford, New York, 2004.
The presence of an extra chromosome 21 leads to a set of traits that, taken together, constitute a syndrome. These traits presumably result from an increase in dosage of the genes and other elements present on chromosome 21, although the specific genes or genetic elements have not been identified. Children with Down syndrome are mentally disabled to varying degrees; most are in the mild to moderate range and with special education and support they can acquire basic communication, self-help, and social skills. People with Down syndrome are short because of delayed maturation of the skeletal system; their muscle tone is low; and they have a characteristic facial appearance. About 40% of children with Down syndrome have major heart defects, and life expectancy is currently about 55 years. Among adults with Down syndrome, the risk of dementia is about 15% to 50%, which is about five times greater than the risk in the general population.
Although Down syndrome affects approximately 1 in 750 live-born children overall, there is a pronounced effect of mother’s age on the risk of Down syndrome. For mothers of ages 45–50, the risk of having a baby with Down syndrome is approximately 50 times greater than it is for mothers of ages 15–20. The increased risk is the reason why many physicians recommend tests to determine abnormalities in the fetus for pregnant women over the age of 35.
Two other trisomies of autosomes (that is, chromosomes other than the X and Y chromosomes), are sometimes found in live births. These are trisomy 13 and trisomy 18. Both are much rarer than Down syndrome and also much more severe. In both cases, the developmental abnormalities are so profound that newborns with these conditions usually do not survive the first year of life. Other trisomies occur, but they are not compatible with life.
15.4.3 Extra sex chromosomes have fewer effects than extra autosomes.
Extra or missing sex chromosomes (X or Y) are also quite common. In most mammals, females have two X chromosomes (XX), and males have one X and one Y chromosome (XY). The presence of the Y chromosome, not the number of X chromosomes, leads to male development. The female karyotype 47, XXX (47 chromosomes, including three X chromosomes) and the male karyotype 47, XYY (47 chromosomes, including one X and two Y chromosomes) are found among healthy females and males. These persons are in the normal range of physical development and mental capability and usually their extra sex chromosome remains undiscovered until their chromosomes are examined for some other reason.
The reason that 47, XYY males show no detectable phenotypic effects is because of the unusual nature of the Y chromosome. The Y contains only a few functional genes other than the gene that stimulates the embryo to take the male developmental pathway. The absence of detectable phenotypic effects in 47, XXX females has a completely different explanation that has to do with the manner in which the activity of genes in the X chromosome is regulated. In the cells of female mammals, all X chromosomes except one are inactivated and gene expression is largely repressed. (This process, called X-inactivation, is discussed in Chapter 20.) Because of X-inactivation, a 47, XXX female has one active X chromosome per cell, the same number as in a normal 46, XX female.
Quick Check 6
A male baby is born with the sex-chromosome constitution XYY. Both parents have normal sex chromosomes (XY in the father, XX in the mother). In which meiotic division of which parent did the nondisjunction take place that produced the XYY baby?
Two other sex-chromosomal abnormalities do have phenotypic effects, especially on sexual development. The more frequent is 47, XXY (47 chromosomes, including two X chromosomes and one Y chromosome). Individuals with this karyotype are male because of the presence of the Y chromosome, but they have a distinctive group of symptoms known as Klinefelter syndrome (Fig. 15.15a). Characteristics of the syndrome are very small testes but normal penis and scrotum. Growth and physical development are, for the most part, normal, although affected individuals tend to be tall for their age. About half of the individuals have some degree of mental impairment. When a person with Klinefelter syndrome reaches puberty, the testes fail to enlarge, the voice remains high pitched, pubic and facial hair remains sparse, and there is some enlargement of the breasts. Affected males do not produce sperm and hence are sterile; up to 10% of men who seek aid at infertility clinics turn out to be XXY.
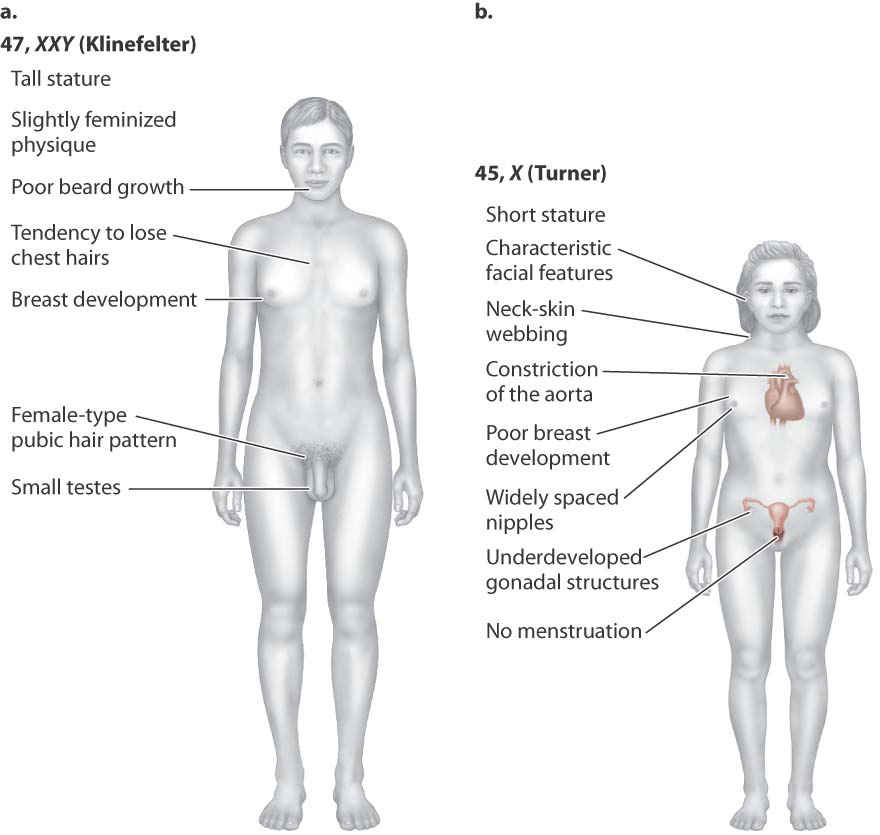
Much rarer than all the other sex-chromosome abnormalities is 45, X (45 chromosomes, with just one X chromosome), the karyotype associated with characteristics known as Turner syndrome (Fig. 15.15b). Affected females are short and often have a distinctive webbing of the skin between the neck and shoulders. There is no sexual maturation. The external sex organs remain immature, the breasts fail to develop, and pubic hair fails to grow. Internally, the ovaries are small or absent, and menstruation does not occur. Although the mental abilities of affected females are very nearly normal, they have specific defects in spatial abilities and arithmetical skills.
Earlier, we saw that in XX females one X chromosome undergoes inactivation. Why, then, do individuals with just one X chromosome show any symptoms at all? The explanation is that some genes on the inactive X chromosome in XX females are not completely inactivated. Evidently, for normal development to take place, both copies of some genes that escape complete inactivation must be expressed.
45, X is one of the rarest karyotypes seen in live-born babies. The reason is that more than 99% of the fetuses that are 45, X undergo spontaneous abortion—the subject of the next section.
15.4.4 Nondisjunction is a major cause of spontaneous abortion.
Trisomies and the sex-chromosome abnormalities discussed in the previous section account for most of the simpler chromosomal abnormalities that are found among babies born alive. However, these chromosomal abnormalities represent only a minority of those that actually occur. Most fertilized eggs with chromosome abnormalities fail to complete embryonic development. At some time during pregnancy—in some cases very early, in other cases relatively late—the chromosomally abnormal embryo or fetus undergoes spontaneous abortion.
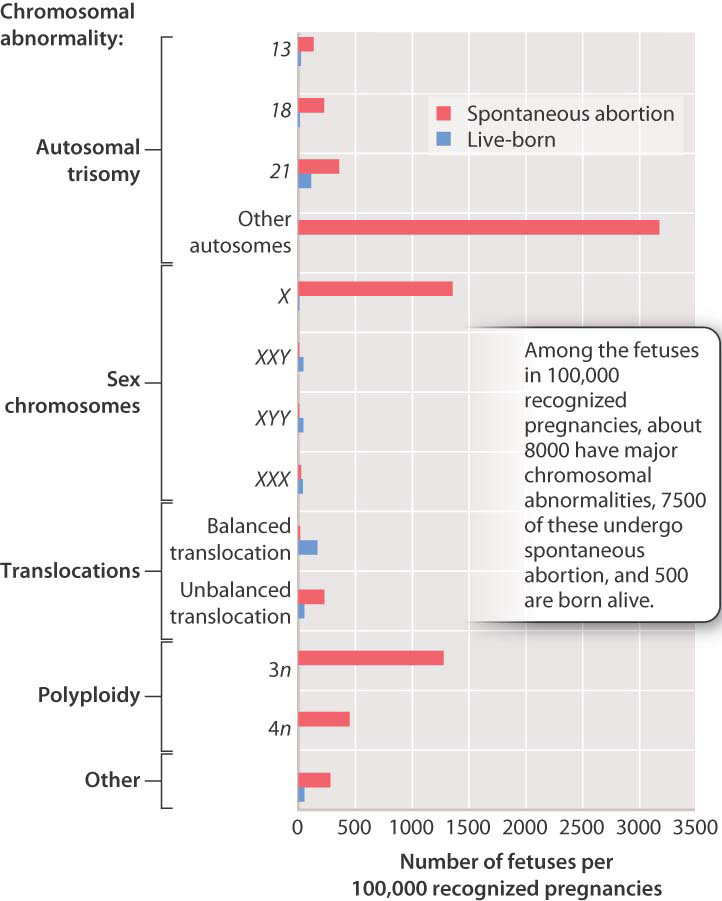
The relative proportions of some of the major chromosomal abnormalities in spontaneous abortion are shown in Fig. 15.16. The bars in red represent recognized pregnancies that terminate in spontaneous abortion, and those in blue represent those in which the fetus develops to term and is born alive. Note the large number of autosomal trisomies, none of which (with the exception of trisomies 13, 18, and 21) permit live births. Even among these, about 75% of fetuses with trisomy 21 undergo spontaneous abortion, and the proportions are even greater for trisomies 13 and 18. The 45, X karyotype is also very frequent among spontaneous abortions.
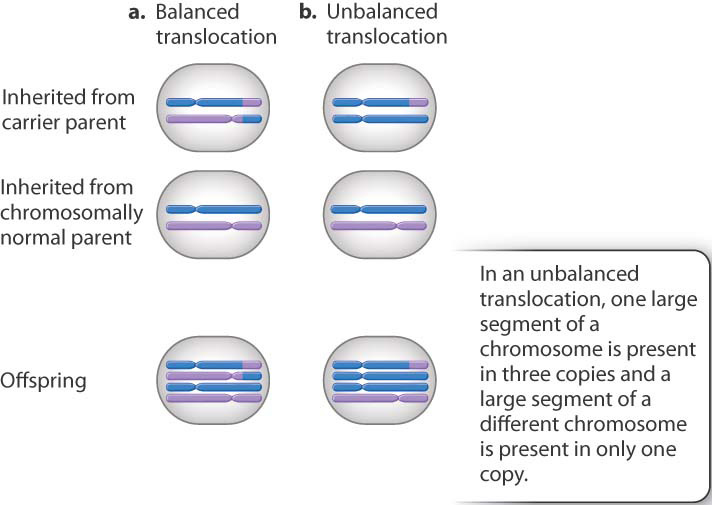
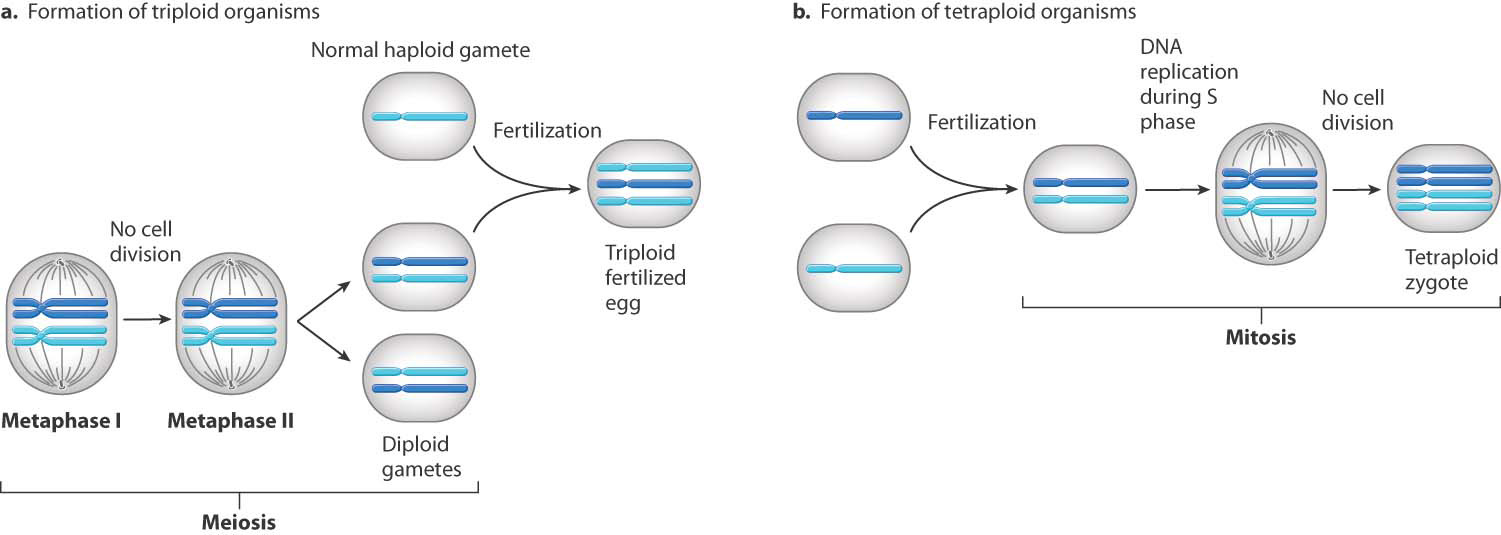
A surprisingly large number of fetuses that undergo spontaneous abortion are triploid (with three complete sets of chromosomes, 69 altogether) or tetraploid (four complete sets, 92 altogether). These karyotypes usually result from a defective spindle apparatus and failure of cell division in anaphase. When this occurs in meiosis, the result is a diploid gamete and a triploid fertilized egg, and when it occurs in the first mitosis in a normal fertilized egg, the result is a tetraploid (Fig. 15.17). Many spontaneous abortions result from an unbalanced translocation (Chapter 14), in which only part of a reciprocal translocation (along with one of the nontranslocated chromosomes) is inherited from one of the parents (Fig. 15.18).
Altogether, about 15% of all recognized pregnancies terminate with spontaneous abortion of the fetus, and roughly half of these are due to major chromosomal abnormalities. This number tells only part of the story because embryos with a missing autosome are not found among spontaneously aborted fetuses. These must occur at least as frequently as those with an extra autosome because both are created by the same event of nondisjunction (see Fig. 15.12). The explanation seems to be that in fertilized eggs with a missing autosome the abortion occurs shortly after fertilization, and most cases are not recognized.