10.4 Determining the Base Sequence of a DNA Segment
After we have cloned and identified our desired gene or have amplified it by PCR, the task of trying to understand its function begins. The ultimate language of the genome is composed of strings of the nucleotides A, T, C, and G. Obtaining the complete nucleotide sequence of a segment of DNA is often an important part of understanding the organization of a gene and its regulation, its relation to other genes, or the function of its encoded RNA or protein. Indeed, the DNA sequence can be used to determine the protein primary structure since, for the most part, translating the nucleic acid sequence of a cDNA molecule to discover the amino sequence of its encoded polypeptide chain is simpler than directly sequencing the polypeptide itself. In this section, we consider the techniques used to read the nucleotide sequence of DNA.
As with recombinant-
This sequencing technique is called dideoxy sequencing or, sometimes, Sanger sequencing after its inventor. The term dideoxy comes from a special modified nucleotide, called a dideoxynucleotide triphosphate (generically, a ddNTP). This modified nucleotide is key to the Sanger technique because of its ability to block continued DNA synthesis. A dideoxynucleotide lacks the 3′-hydroxyl group as well as the 2′-hydroxyl group, which is also absent in a deoxynucleotide (Figure 10-16; compare to Figure 7-
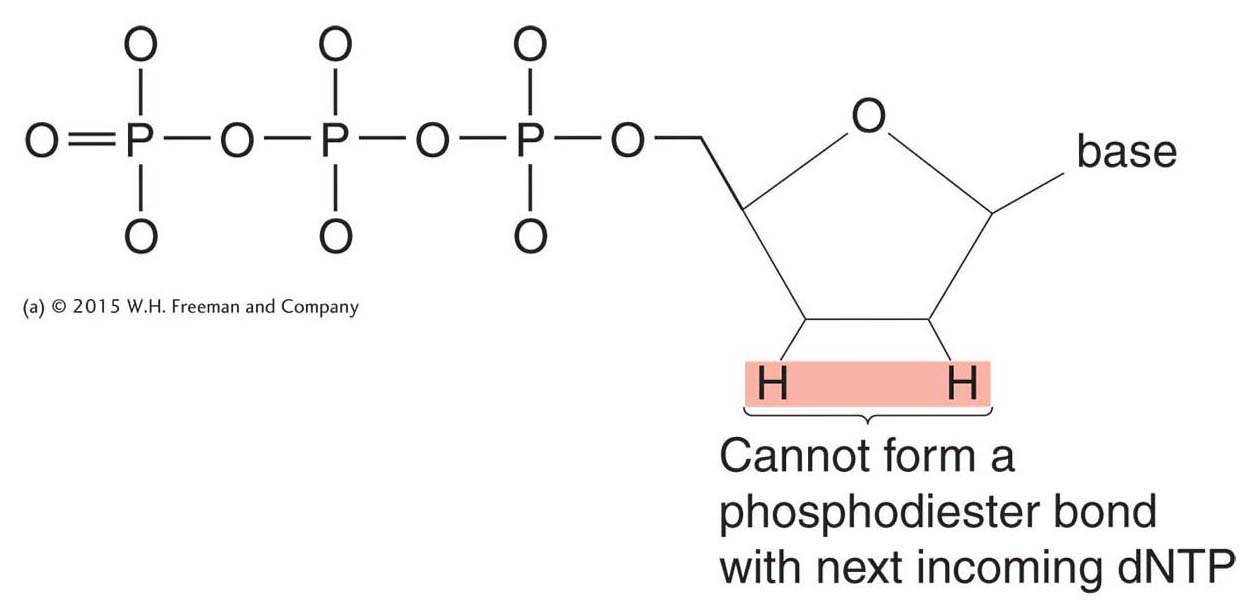
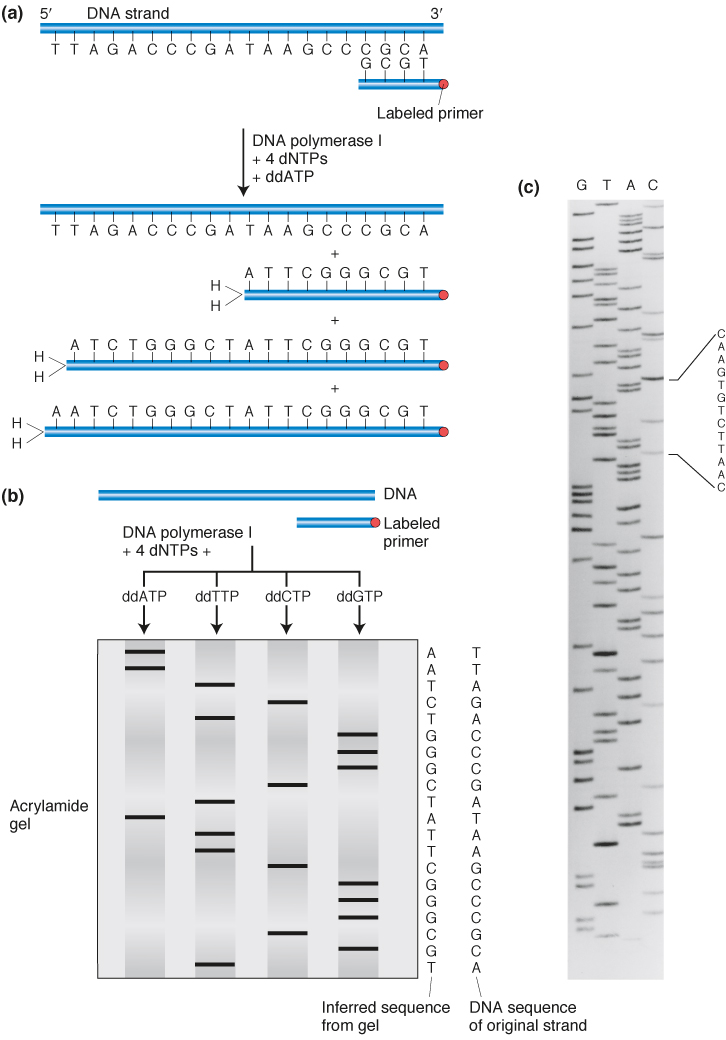
The logic of dideoxy sequencing is straightforward. Suppose we want to read the sequence of a cloned DNA segment of up to 800 base pairs. This DNA segment could be a plasmid insert or even a PCR product. First, we denature the two strands of this segment. Next, we create a primer for DNA synthesis that will hybridize to exactly one location on the cloned DNA segment and then add a special “cocktail” of DNA polymerase, normal deoxynucleotide triphosphates (dATP, dCTP, dGTP, and dTTP), and a small amount of a dideoxynucleotide for one of the four bases (for example, ddATP). The polymerase will begin to synthesize the complementary DNA strand, starting from the primer, but will stop at any point at which the dideoxynucleotide triphosphate is incorporated into the growing DNA chain in place of the normal deoxynucleotide triphosphate. Suppose the DNA segment that we’re trying to sequence is
375
5′ ACGGGATAGCTAATTGTTTACCGCCGGAGCCA 3′
We would then start DNA synthesis from a complementary primer:
Using the special DNA-

We can generate an array of such fragments for each of the four possible dideoxynucleotide triphosphates in four separate cocktails (one spiked with ddATP, one with ddCTP, one with ddGTP, and one with ddTTP). Each will produce a different array of fragments, with no two spiked cocktails producing fragments of the same size. Next, the DNA fragments generated in the four cocktails are separated and displayed in order using gel electrophoresis. By running the fragments in four adjacent lanes of a polyacrylamide gel that can resolve fragments of DNA that vary by only one nucleotide in length, we see that the fragments can be ordered by length with the lengths increasing by one base at a time.
The newly synthesized strands must be labeled in some way to make the bands visible on the gel. Strands are labeled as they are made either by using a primer that is radioactively labeled or having one of the regular dNTPs carry a radioactive label. Fluorescent labels can also be used, and in this case they are carried by each ddNTP (see below).
The products of such dideoxy sequencing reactions are shown in Figure 10-17. That result is a ladder of labeled DNA chains increasing in length by one, and so all we need do is read up the gel to read the DNA sequence of the synthesized strand in the 5′-to-
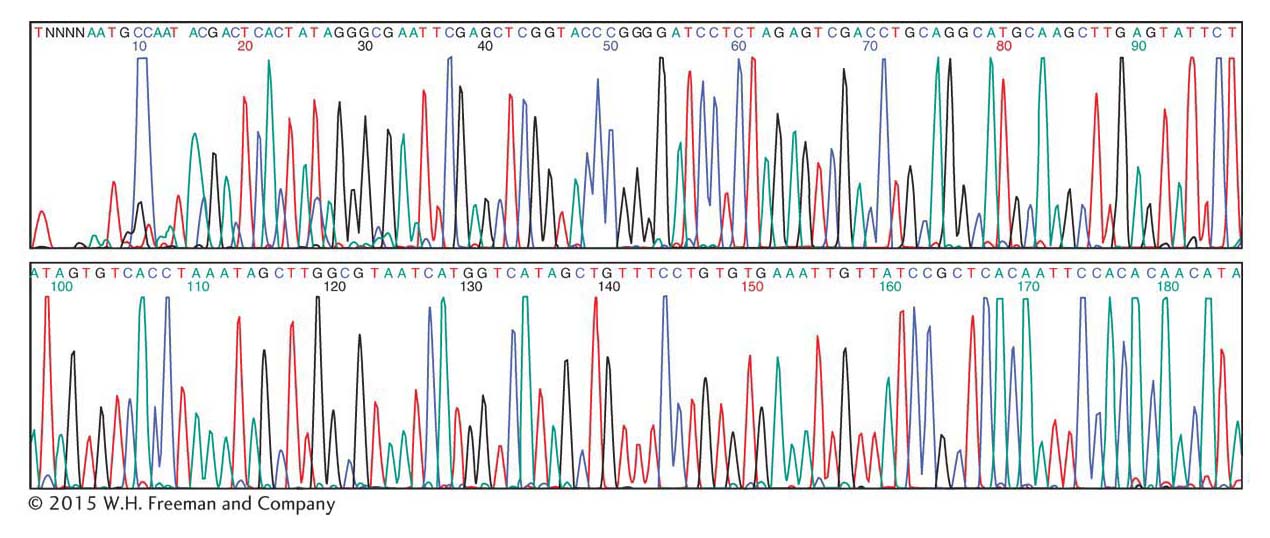
If the tag is a fluorescent dye, a different fluorescent color emitter is used for each of the four ddNTP reactions, and a detector at the end of the gel can distinguish each color. The four reactions take place in the same test tube, and the four sets of nested DNA chains can undergo electrophoresis together. Thus, four times as many sequences can be produced in the same amount of time as can be produced by running the reactions separately. This logic is used in fluorescence detection by automated DNA-
376
377