PROBLEMS
WORKING WITH THE FIGURES
Question 1
Based on Figure 14-2, why must the DNA fragments sequenced overlap in order to obtain a genome sequence?
Question 2
Filling gaps in draft genome sequences is a major challenge. Based on Figure 14-6, can paired-
Question 3
In Figure 14-9, how are the positions of codons determined?
Question 4
In Figure 14-10, expressed sequence tags (ESTs) are aligned with genomic sequence. How are ESTs helpful in genome annotation?
Question 5
In Figure 14-10, cDNA sequences are aligned with genomic sequence. How are cDNA sequences helpful in genome annotation? Are cDNAs more important for bacterial or eukaryotic genome annotations?
Question 6
Based on Figure 14-14 and the features of ultraconserved elements, what would you predict you’d observe if you injected a reporter-
Question 7
Figure 14-17 shows syntenic regions of mouse chromosome 11 and human chromosome 17. What do these syntenic regions reveal about the genome of the last common ancestor of mice and humans?
Question 8
In Figure 14-18, what key step enables exome sequencing and distinguishes it from whole-
Question 9
The genomes of two E. coli strains are compared in Figure 14-19. Would you expect any third strain to contain more of the blue, tan, or red regions shown in Figure 14-19? Explain.
Question 10
Figure 14-21 depicts the Gal4-
BASIC PROBLEMS
Question 11
Explain the approach that you would apply to sequencing the genome of a newly discovered bacterial species.
Question 12
Terminal-
Question 13
What is the difference between a contig and a scaffold?
Question 14
Two particular contigs are suspected to be adjacent, possibly separated by repetitive DNA. In an attempt to link them, end sequences are used as primers to try to bridge the gap. Is this approach reasonable? In what situation will it not work?
Question 15
A segment of cloned DNA containing a protein-
Question 16
In an in situ hybridization experiment, a certain clone bound to only the X chromosome in a boy with no disease symptoms. However, in a boy with Duchenne muscular dystrophy (X-
Question 17
In a genomic analysis looking for a specific disease gene, one candidate gene was found to have a single-
Question 18
Is a bacterial operator a binding site?
Question 19
A certain cDNA of size 2 kb hybridized to eight genomic fragments of total size 30 kb and contained two short ESTs. The ESTs were also found in two of the genomic fragments each of size 2 kb. Sketch a possible explanation for these results.
Question 20
A sequenced fragment of DNA in Drosophila was used in a BLAST search. The best (closest) match was to a kinase gene from Neurospora. Does this match mean that the Drosophila sequence contains a kinase gene?
Question 21
In a two-
Question 22
You have the following sequence reads from a genomic clone of the Drosophila melanogaster genome:
Read 1: TGGCCGTGATGGGCAGTTCCGGTG
Read 2: TTCCGGTGCCGGAAAGA
Read 3: CTATCCGGGCGAACTTTTGGCCG
Read 4: CGTGATGGGCAGTTCCGGTG
Read 5: TTGGCCGTGATGGGCAGTT
Read 6: CGAACTTTTGGCCGTGATGGGCAGTTCC
Use these six sequence reads to create a sequence contig of this part of the D. melanogaster genome.
Question 23
Sometimes, cDNAs turn out to be “chimeras”; that is, fusions of DNA copies of two different mRNAs accidentally inserted adjacently to each other in the same clone. You suspect that a cDNA clone from the nematode Caenorhabditis elegans is such a chimera because the sequence of the cDNA insert predicts a protein with two structural domains not normally observed in the same protein. How would you use the availability of the entire genomic sequence to assess if this cDNA clone is a monster or not?
Question 24
In browsing through the human genome sequence, you identify a gene that has an apparently long coding region, but there is a two-
How would you determine whether the deletion was correct or an error in the sequencing?
You find that the exact same deletion exists in the chimpanzee homolog of the gene but that the gorilla gene reading frame is intact. Given the phylogeny of great apes below, what can you conclude about when in ape evolution the mutation occurred?
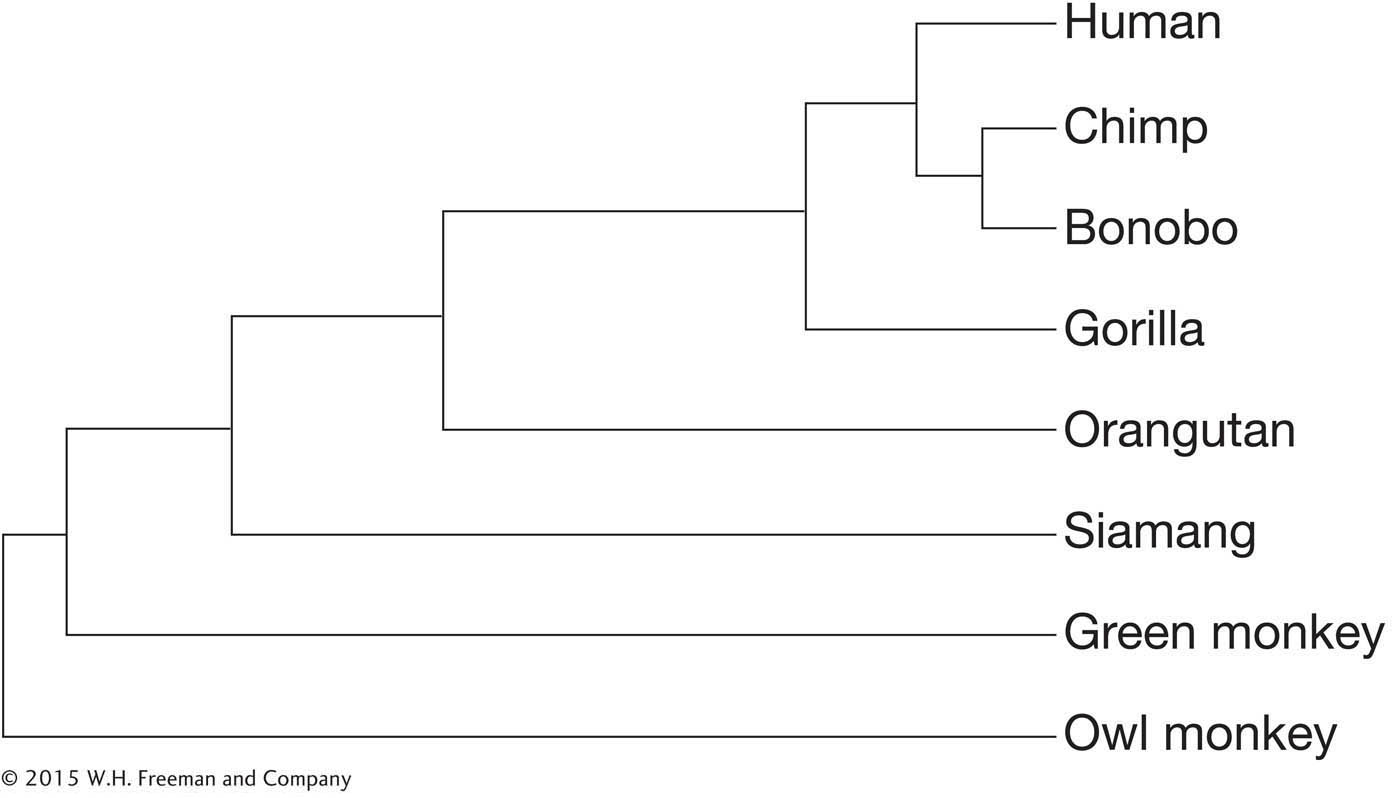
Question 25
In browsing through the chimpanzee genome, you find that it has three homologs of a particular gene, whereas humans have only two.
What are two alternative explanations for this observation?
How could you distinguish between these two possibilities?
Question 26
The platypus is one of the few venomous mammals. The male platypus has a spur on the hind foot through which it can deliver a mixture of venom proteins. Looking at the phylogeny in Figure 14-15, how would you go about determining whether these venom proteins are unique to the platypus?
Question 27
You have sequenced the genome of the bacterium Salmonella typhimurium, and you are using BLAST analysis to identify similarities within the S. typhimurium genome to known proteins. You find a protein that is 100 percent identical in the bacterium Escherichia coli. When you compare nucleotide sequences of the S. typhimurium and E. coli genes, you find that their nucleotide sequences are only 87 percent identical.
Explain this observation.
What do these observations tell you about the merits of nucleotide-
versus protein- similarity searches in identifying related genes?
Question 28
To inactivate a gene by RNAi, what information do you need? Do you need the map position of the target gene?
Question 29
What is the purpose of generating a phenocopy?
Question 30
What is the difference between forward and reverse genetics?
Question 31
Why might exome sequencing fail to identify a disease-
CHALLENGING PROBLEMS
Question 32
You have the following sequence reads from a genomic clone of the Homo sapiens genome:
Read 1: ATGCGATCTGTGAGCCGAGTCTTTA
Read 2: AACAAAAATGTTGTTATTTTTATTTCAGATG
Read 3: TTCAGATGCGATCTGTGAGCCGAG
Read 4: TGTCTGCCATTCTTAAAAACAAAAATGT
Read 5: TGTTATTTTTATTTCAGATGCGA
Read 6: AACAAAAATGTTGTTATT
Use these six sequence reads to create a sequence contig of this part of the H. sapiens genome.
Translate the sequence contig in all possible reading frames.
Go to the BLAST page of the National Center for Biotechnology Information, or NCBI and see if you can identify the gene of which this sequence is a part by using each of the reading frames as a query for protein–
protein comparison (BLASTp).
Question 33
Some sizable regions of different chromosomes of the human genome are more than 99 percent nucleotide identical with one another. These regions were overlooked in the production of the draft genome sequence of the human genome because of their high level of similarity. Of the techniques discussed in this chapter, which would allow genome researchers to identify the existence of such duplicate regions?
Question 34
Some exons in the human genome are quite small (less than 75 bp long). Identification of such “microexons” is difficult because these distances are too short to reliably use ORF identification or codon bias to determine if small genomic sequences are truly part of an mRNA and a polypeptide. What techniques of “gene finding” can be used to try to assess if a given region of 75 bp constitutes an exon?
Question 35
You are studying proteins having roles in translation in the mouse. By BLAST analysis of the predicted proteins of the mouse genome, you identify a set of mouse genes that encode proteins with sequences similar to those of known eukaryotic translation-
Would you use forward-
or reverse- genetics approaches to identify these mutations? Briefly outline two different approaches that you might use to look for loss-
of- function phenotypes in one of these genes.
Question 36
The entire genome of the yeast Saccharomyces cerevisiae has been sequenced. This sequencing has led to the identification of all the open reading frames (ORFs, gene-
15 percent are lethal when knocked out.
25 percent show some mutant phenotype (altered morphology, altered nutrition, and so forth).
60 percent show no detectable mutant phenotype at all and resemble wild type.
Explain the possible molecular-
Question 37
Different strains of E. coli are responsible for enterohemorrhagic and urinary tract infections. Based on the differences between the benign K-
between K-
12 and uropathogenic strains? between O157:H7 and uropathogenic strains?
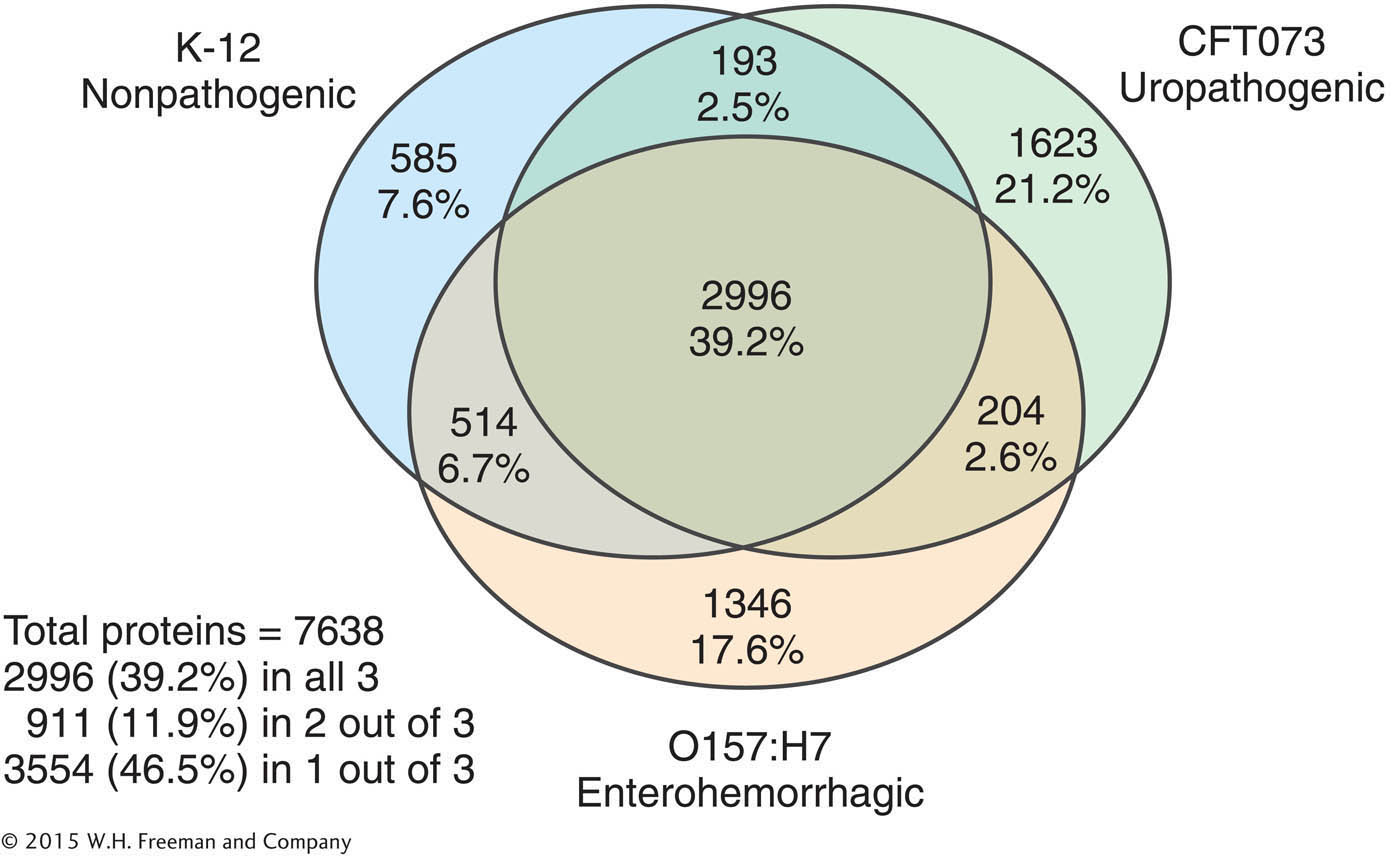
What might explain the observed pair-
by- pair differences in genome content? How might the function of strain-
specific genes be tested?