14.6 Comparative Genomics and Human Medicine
The human species, Homo sapiens, originated in Africa approximately 200,000 years ago. Around 60,000 years ago, populations left Africa and migrated across the world, eventually populating five additional continents. These migrating populations encountered different climates, adopted different diets, and combated different pathogens in different parts of the world. Much of the recent evolutionary history of our species is recorded in our genomes, as are the genetic differences that make individuals or populations more or less susceptible to disease.
Overall, any two unrelated humans’ genomes are 99.9 percent identical. That difference of just 0.1 percent still corresponds to roughly 3 million bases. The challenge today is to decipher which of those base differences are meaningful with respect to physiology, development, or disease.
Once the sequence of the first human genome was advanced, that accomplishment opened the door to much more rapid and less costly analysis of other individuals. The reason is that with a known genome assembly as a reference, it is much easier to align the raw sequence reads of additional individuals, and to design approaches to studying and comparing parts of the genome.
One of the first and greatest surprises that has emerged from comparing individual human genomes is that humans differ not merely at one base in a thousand, but also in the number of copies of parts of individual genes, entire genes, or sets of genes. These copy number variations (CNVs) include repeats and duplications that increase copy number and deletions that reduce copy number. Between any two unrelated individuals, there may be 1000 or more segments of DNA greater than 500 bp in length that differ in copy number. Some CNVs can be quite large and span over 1 million base pairs.
How such copy numbers may play a role in human evolution and disease is of intense interest. One case where increased copy number appears to have been adaptive concerns diet. People with high-
The exome and personalized genomics
WHAT GENETICISTS ARE DOING TODAY
Advances in sequencing technologies have reduced the cost of sequencing individual genomes from about $300 million in 2000, to $1 million in 2008, to about $5000 in 2013. But for many large-
The strategy for exome sequencing involves generating a library of genomic DNA that is enriched for exon sequences (Figure 14-18). The DNA is prepared by (1) shearing genomic DNA into short, single-
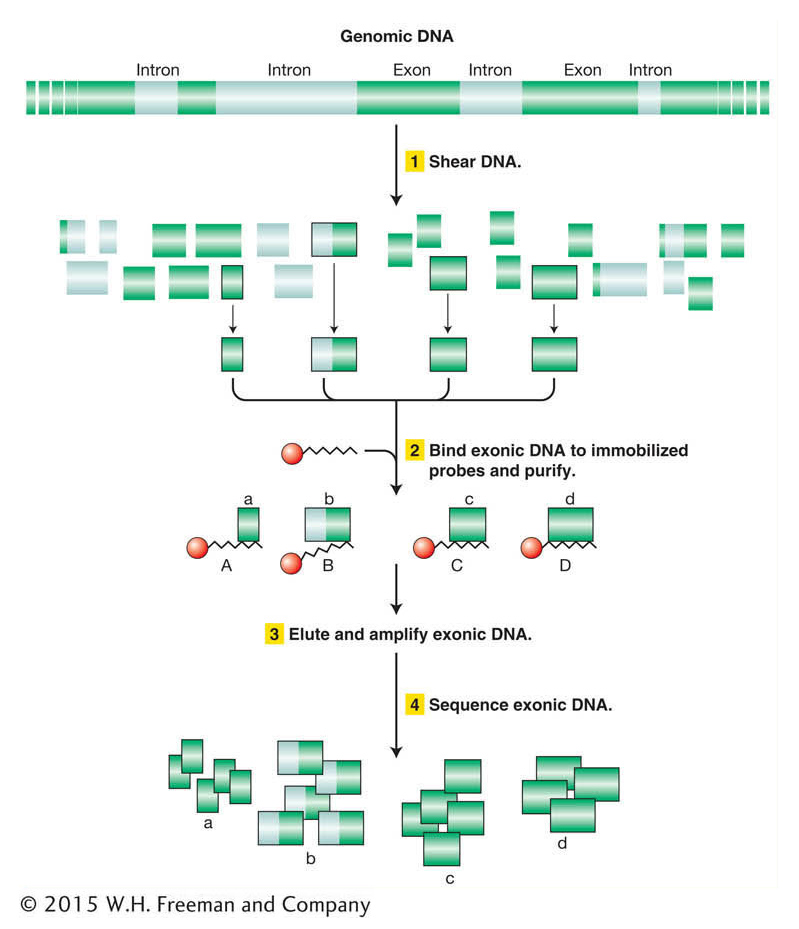
As of late 2013, the exomes of more than 100,000 individuals have been sequenced, at the current cost of only ~$500 per exome. One particularly important power of exome sequencing is to identify de novo mutations in individuals (mutations that are not present in either parent). Such mutations are responsible for many spontaneously appearing genetic diseases whose origins would not be revealed by traditional pedigree-
And just as exome sequencing can be used to identify genetic differences between individuals, it can also be used to identify differences between normal and abnormal cells, such as cancer cells. Cancer is a suite of genetic diseases in which combinations of gene mutations typically contribute to the loss of growth control and metastasis. Understanding what genetic changes are common to particular cancers, or to subsets of cancers, will not only further our understanding of cancer, but also promises to impact diagnosis and treatment in powerful ways. Researchers across the world are collaborating to create an “atlas” of cancer genomes that compiles our expanding knowledge of the genetic mutations associated with many cancers. (See http:/
The ability to rapidly analyze organisms’ genomes is also impacting other dimensions of medicine. We will look at one such case next.
Comparative genomics of nonpathogenic and pathogenic E. coli
Escherichia coli are found in our mouths and intestinal tracts in vast numbers, and this species is generally a benign symbiont. Because of its central role in genetics research, it was one of the first bacterial genomes sequenced. The E. coli genome is about 4.6 Mb in size and contains 4405 genes. However, calling it “the E. coli genome” is really not accurate. The first genome sequenced was derived from the common laboratory E. coli strain K-
In 1982, a multistate outbreak of human disease was traced to the consumption of undercooked ground beef. The E. coli strain O157:H7 was identified as the culprit, and it has since been associated with a number of large-
To understand the genetic bases of pathogenicity, the genome of an E. coli O157:H7 strain has been sequenced. The O157 and K-
Despite the similarities in many proteins, the genomes and proteomes differ enormously in content. The E. coli O157 genome encodes 5416 genes, whereas the E. coli K-
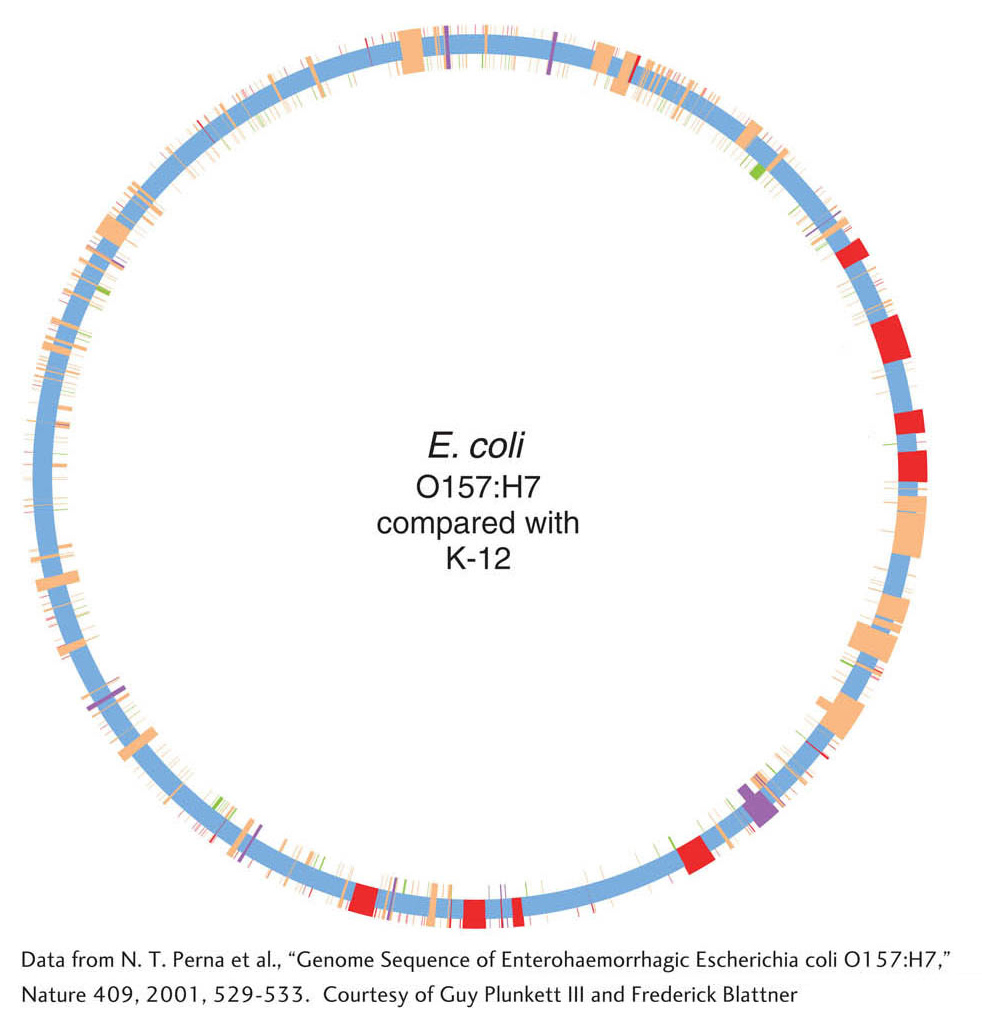
Among the 1387 genes specific to E. coli O157 are many genes that are suspected to encode virulence factors, including toxins, cell-
The surprising level of diversity between two members of the same species shows how dynamic genome evolution can be. Most new genes in E. coli strains are thought to have been introduced by horizontal transfer from the genomes of viruses and other bacteria (see Chapter 5). Differences can also evolve owing to gene deletion. Other pathogenic E. coli and bacterial species also exhibit many differences in gene content from their nonpathogenic cousins. The identification of genes that may contribute directly to pathogenicity opens new avenues to the understanding, prevention, and treatment of infectious disease.