Chapter 5. Polyglutamine diseases: from expansion mutations to targets for therapy
Introduction
Polyglutamine diseases: from expansion mutations to targets for therapy
Introduction
Certain genes contain sequences of the three nucleotides cytosine-
Polyglutamine toxicity might also contribute to other trinucleotide expansion diseases. For example, in the neurodegenerative disease spinocerebellar ataxia 8 (SCA8), which was found to be caused by a CTG repeat expansion, animal studies have shown evidence for transcription of the opposite strand, where CTG is read as CAG2. This bidirectional expression may also apply to other trinucleotide repeat disorders, such as myotonic dystrophy (DM1)3 and Huntington disease–
1.La Spada, A. R. et al. Androgen receptor gene mutations in X-
2. Moseley, M. L. et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nature Genetics 38, 758–
3.Cho, D. H. et al. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Molecular Cell 20, 483–
4.Wilburn, B. et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-
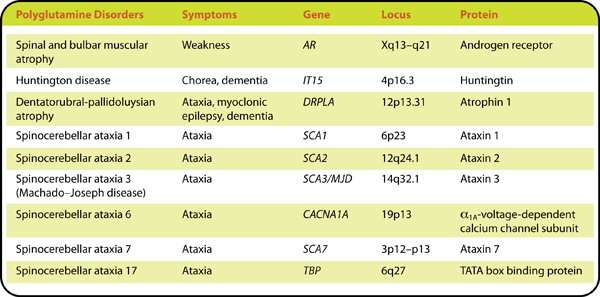
The Polyglutamine Diseases
Although the genes associated with SBMA, HD, DRPLA, and SCA types 1, 2, 3, 6, 7, and 17 are structurally and functionally distinct, these polyglutamine diseases share certain key characteristics. They are all progressive, often fatal disorders that typically begin in adulthood and worsen over a period of 10 to 30 years. The diseases occur only when the length of the repeat exceeds a threshold number, ranging from about 20 to 50 CAGs. With the exception of SBMA, which is X-
Individuals with longer CAG repeats have more severe symptoms and disease onset earlier in life. For example, in HD, the median age of onset decreases from 67 years for patients with 39 CAGs to 27 years for patients with 50 CAGs5. Furthermore, the severity of the disease may increase from one generation to the next because the length of the CAG repeat is unstable and tends to increase with transmission. This phenomenon is known as “anticipation.”
How can this tendency toward expansion of the CAG repeats be explained? The characteristic instability of repeats can be caused by mistakes that occur during DNA replication, including strand slippage, misalignment, and stalling (Figure 1)6. Single-
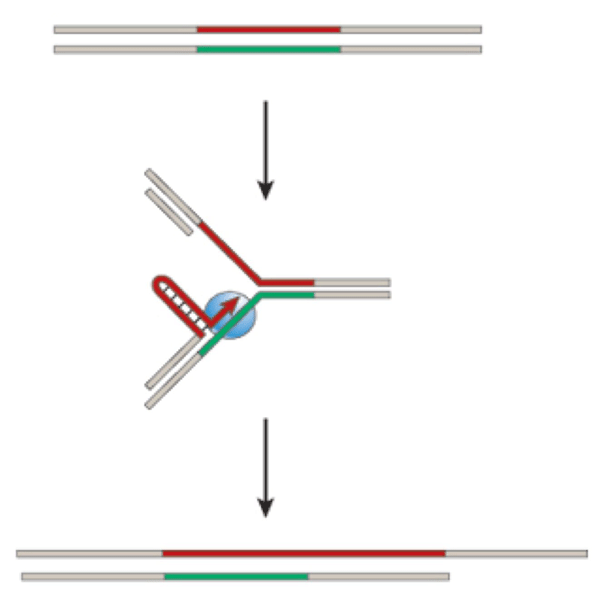
Figure 1 Source: Modified version of Figure 3 from Nature 447, 932–
5.Brinkman, R. R. et al. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. American Journal of Human Genetics 60, 1202–
6.Mirkin, S. M. Expandable DNA repeats and human disease. Nature 447, 932–
Protein Aggregation
Expanded polyglutamine tracts are sticky and make the disease-
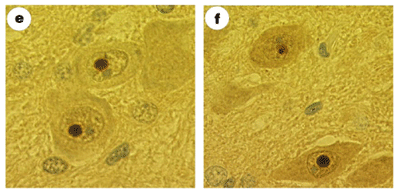
Figure 2 Source: Figure 1 (panels e and f only) from Skinner, P. J. et al. Ataxin-
7.DiFiglia, M. et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–
8.Davies, S. W. et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–
9.Paulson, H. L. et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19, 333–
10. Klement, I. A. et al. Ataxin-
11. Saudou, F. et al. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–
12. Arrasate, M. et al. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–
13. Hackam, A. S. et al. The influence of huntingtin protein size on nuclear localization and cellular toxicity. Journal of Cell Biology 141, 1097–
How do Polyglutamine Disease Proteins Cause Cell Damage?
Many hypotheses have been put forward to explain the mechanism of polyglutamine disease (Figure 3). Here are some of the more likely possibilities.
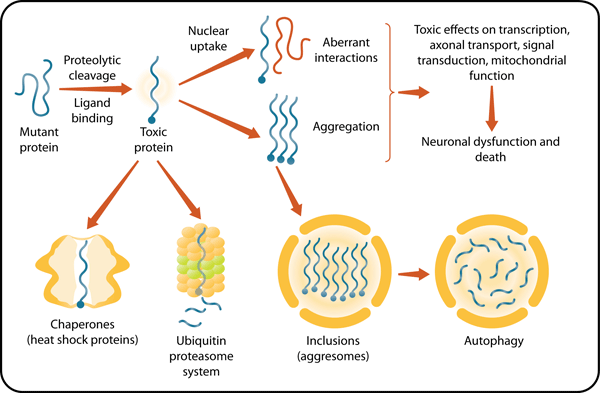
Figure 3 Source: Original designed by author and illustrator. Published previously in Pennuto, M. & Fischbeck, K. H. Therapeutic prospects for polyglutamine disease. In Ramirez-
Transcriptional Dysregulation
One way to explain the toxic effects of polyglutamine proteins is that they accumulate in the nucleus and have abnormal interactions with nuclear proteins, which in turn disrupt transcription14. Many proteins that normally regulate transcription are found in nuclear inclusions and have been shown to interact with polyglutamine expanded proteins15. The mutant proteins may prevent these transcriptional regulators from carrying out their normal functions.
14.Zoghbi, H. Y . & Orr, H. T. Glutamine repeats and neurodegeneration. Annual Review of Neuroscience 23, 217–
15.Yamada, M., Tsuji, S. & Takahashi, H. Pathology of CAG repeat diseases. Neuropathology 20, 319–
Mitochondrial Dysfunction
Several findings show that polyglutamine diseases are associated with mitochondrial dysfunction. For example, mitochondria isolated from HD patients and a mouse model of HD have defects in their membrane potential and depolarization16. Mutant forms of huntingtin may disrupt mitochondrial function by decreasing the expression of the transcriptional coactivator PGC-
16.Panov, A. V. et al. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nature Neuroscience 5, 731–
17.Cui, L. et al. Transcriptional repression of PGC-
Impairment of the Ubiquitin-Proteasome System
The proteasome is a protein complex that degrades unneeded or damaged proteins. Proteins are targeted for proteasome-
9.Paulson, H. L. et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19, 333–
18.Chai, Y. et al. Evidence for proteasome involvement in polyglutamine disease: Localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Human Molecular Genetics 8, 673–
19.Burnett, B., Li, F. & Pittman, R. N. The polyglutamine neurodegenerative protein ataxin-
20.Chai, Y. et al. Poly-
21.Donaldson, K. M. et al. Ubiquitin-
22.Bowman, A. B. et al. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-
23.Bett, J. S. et al. The ubiquitin-
The Role of Autophagy
Autophagy is a process by which cellular components, including damaged or surplus organelles and protein aggregates, are delivered to the lysosome for degradation. In humans, defects in autophagy have been directly associated with neurodegeneration, cancer, and inflammatory diseases24. Signs of decreased efficiency of autophagy have been reported in several polyglutamine diseases, which may be contributing to the disease mechanism25.
24.Kundu, M. & Thompson, C. B. Autophagy: Basic principles and relevance to disease. Annual Review of Pathology 3, 427–
25.Martinez-
Other Mechanisms
Other cellular processes have been implicated in the toxicity of mutant polyglutamine proteins. For example, altered cellular signaling, intracellular calcium homeostasis26, 27, and axonal transport28, 29 have been found in several polyglutamine disorders.
26.Zeron, M. M. et al. Increased sensitivity to N-
27.Tang, T. S. et al. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proceedings of the National Academy of Sciences of the United States of America 102, 2602–
28.Gunawardena, S. et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40, 25–
29.Szebenyi, G. et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40, 41–
The Protein Context
Although long polyglutamine tracts alone may be toxic and cause pathology in cell culture and animal models30, 31, the protein context is also important. For example, the toxicity of the mutant AR is dependent on the binding of androgens in animal models of SBMA32, and transgenic mice with high expression levels of the wild-
Protein context is also important in the polyglutamine disease SCA1, which is associated with the ataxin-
Polyglutamine expansions may enhance normal or toxic activities associated with certain conformations of the protein (causing gain of function) and weaken the activities associated with others (causing loss of function). For example, a combination of loss and gain of function explains the clinical picture of SBMA, where patients often have signs of androgen insensitivity (breast enlargement and reduced fertility) combined with signs of motor neuron degeneration (weakness and muscle atrophy) due to acquired functions of the AR protein that are toxic to motor neurons.
30.Ordway, J. M. et al. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 91, 753–
31.Marsh, J. L. et al. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Human Molecular Genetics 9, 13–
32.Katsuno, M. et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35, 843–
33.Monks, D. A. et al. Overexpression of wild-
34.Emamian, E. S. et al. Serine 776 of ataxin-
35.Duvick, L. et al. SCA1-
36.Gu, X. et al. Serines 13 and 16 are critical determinants of full-
SUMMARY: Unanswered Questions and Routes to Therapy
Our understanding of the mechanisms underlying polyglutamine diseases has broadened over the last 20 years. This increased understanding has allowed the identification of potential therapeutic targets—
Our understanding of polyglutamine disease mechanisms helps us design assays to identify compounds that can be tested in disease-
References
1.La Spada, A. R. et al. Androgen receptor gene mutations in X-
2. Moseley, M. L. et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nature Genetics 38, 758–
3.Cho, D. H. et al. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Molecular Cell 20, 483–
4.Wilburn, B. et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-
5.Brinkman, R. R. et al. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. American Journal of Human Genetics 60, 1202–
6.Mirkin, S. M. Expandable DNA repeats and human disease. Nature 447, 932–
7.DiFiglia, M. et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–
8.Davies, S. W. et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–
9.Paulson, H. L. et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 19, 333–
10. Klement, I. A. et al. Ataxin-
11. Saudou, F. et al. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–
12. Arrasate, M. et al. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–
13. Hackam, A. S. et al. The influence of huntingtin protein size on nuclear localization and cellular toxicity. Journal of Cell Biology 141, 1097–
14.Zoghbi, H. Y . & Orr, H. T. Glutamine repeats and neurodegeneration. Annual Review of Neuroscience 23, 217–
15.Yamada, M., Tsuji, S. & Takahashi, H. Pathology of CAG repeat diseases. Neuropathology 20, 319–
16.Panov, A. V. et al. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nature Neuroscience 5, 731–
17.Cui, L. et al. Transcriptional repression of PGC-
18.Chai, Y. et al. Evidence for proteasome involvement in polyglutamine disease: Localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Human Molecular Genetics 8, 673–
19.Burnett, B., Li, F. & Pittman, R. N. The polyglutamine neurodegenerative protein ataxin-
20.Chai, Y. et al. Poly-
21.Donaldson, K. M. et al. Ubiquitin-
22.Bowman, A. B. et al. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-
23.Bett, J. S. et al. The ubiquitin-
24.Kundu, M. & Thompson, C. B. Autophagy: Basic principles and relevance to disease. Annual Review of Pathology 3, 427–
25.Martinez-
26.Zeron, M. M. et al. Increased sensitivity to N-
27.Tang, T. S. et al. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proceedings of the National Academy of Sciences of the United States of America 102, 2602–
28.Gunawardena, S. et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40, 25–
29.Szebenyi, G. et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40, 41–
30.Ordway, J. M. et al. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 91, 753–
31.Marsh, J. L. et al. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Human Molecular Genetics 9, 13–
32.Katsuno, M. et al. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35, 843–
33.Monks, D. A. et al. Overexpression of wild-
34.Emamian, E. S. et al. Serine 776 of ataxin-
35.Duvick, L. et al. SCA1-
36.Gu, X. et al. Serines 13 and 16 are critical determinants of full-
37.Palazzolo, I. et al. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Human Molecular Genetics 16, 1593–
Quiz
Question 5.1
1. Which of the following statements could explain why there is often a threshold number of polyglutamine repeats necessary before a disease state begins?
| A. |
| B. |
| C. |
| D. |
Question 5.2
2. Which of the following results would NOT suggest that aggregation causes disease?
| A. |
| B. |
| C. |
| D. |
Question 5.3
3. Which result is consistent with the model that the ubiquitin-proteasome system is involved in spinocerebellar ataxia type 7 (SCA7) neurodegenerative disease?
| A. |
| B. |
| C. |
| D. |
Question 5.4
4. How might polyglutamine expansions cause loss of function to the resulting protein?
| A. |
| B. |
| C. |
| D. |
Question 5.5
5. Which of the following statements is NOT a reason that it is feasible to screen for drugs and develop treatments for polyglutamine diseases?
| A. |
| B. |
| C. |
| D. |
Question 5.6
6. How might polyglutamine proteins affect transcription?
| A. |
| B. |
| C. |
| D. |
Question 5.7
7. How does transcription contribute to the formation of polyglutamine expansions?
| A. |
| B. |
| C. |
| D. |