SR Proteins Contribute to Exon Definition in Long Pre-mRNAs
The average length of an exon in the human genome is about 150 bases, whereas the average length of an intron is about 3500 bases, and the longest introns exceed 500 kb! Because the sequences of 5′ and 3′ splice sites and branch points are so degenerate, multiple copies of those sequences are likely to occur randomly in long introns. Consequently, additional sequence information is required to define the exons that should be spliced together in higher organisms with long introns.
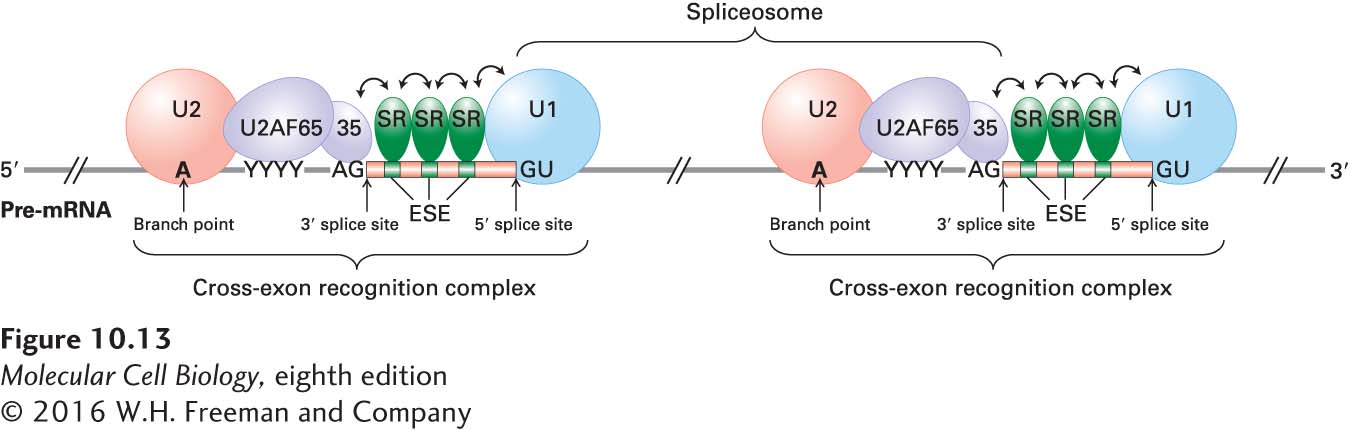
The information for defining the splice sites that demarcate exons is encoded within the sequences of the exons. A family of RNA-binding proteins, the SR proteins, interact with sequences within exons called exonic splicing enhancers. SR proteins are a subset of the hnRNP proteins discussed earlier that contain one or more RRM RNA-binding domains. They also contain several protein-protein interaction domains rich in arginine (R) and serine (S) residues, called RS domains. When bound to exonic splicing enhancers, SR proteins mediate the cooperative binding of U1 snRNP to a true 5′ splice site and U2 snRNP to a branch point through a network of protein-protein interactions that span an exon (Figure 10-13). The complex of SR proteins, snRNPs, and other splicing factors (e.g., U2AF and SF1) that assemble across an exon, which has been called a cross-exon recognition complex, permits precise specification of exons in long pre-mRNAs.
FIGURE 10-13Exon recognition through cooperative binding of SR proteins and splicing factors to pre-mRNA. The correct 5′ GU and 3′ AG splice sites are recognized by splicing factors on the basis of their proximity to exons. The exons contain exonic splicing enhancers (ESEs) that are binding sites for SR proteins. When bound to ESEs, the SR proteins interact with one another and promote the cooperative binding of the U1 snRNP to the 5′ splice site of the downstream intron, SF1 and then the U2 snRNP to the branch point of the upstream intron, the 65- and 35-kDa subunits of U2AF to the polypyrimidine tract and AG 3′ splice site of the upstream intron, and other splicing factors (not shown). The resulting RNA-protein cross-exon recognition complex spans an exon and activates the correct splice sites for RNA splicing. Note that the U1 and U2 snRNPs in this unit do not become part of the same spliceosome. The U2 snRNP on the right forms a spliceosome with the U1 snRNP bound to the 5′ end of the same intron. The U1 snRNP shown on the right forms a spliceosome with the U2 snRNP bound to the branch point of the downstream intron (not shown), and the U2 snRNP on the left forms a spliceosome with a U1 snRNP bound to the 5′ splice site of the upstream intron (not shown). Double-headed arrows indicate protein-protein interactions. See T. Maniatis, 2002, Nature418:236; see also S. M. Berget, 1995, J. Biol. Chem.270:2411.
Mutations that interfere with the binding of an SR protein to an exonic splicing enhancer, even if they do not change the encoded amino acid sequence, prevent formation of the cross-exon recognition complex. As a result, the affected exon is “skipped” during splicing and is not included in the final processed mRNA. The truncated mRNA produced in this case is either degraded or translated into a mutant, abnormally functioning protein. This type of mutation occurs in some human genetic diseases. For example, spinal muscular atrophy is one of the most common genetic causes of childhood mortality. This disease results from mutations in a region of the genome containing two closely related genes, SMN1 and SMN2, that arose by gene duplication. The two genes encode identical proteins, but SMN2 is expressed at a much lower level because a silent mutation in one exon interferes with the binding of an SR protein. This mutation leads to exon skipping in most of the SMN2 mRNAs. The homologous SMN gene in the mouse, in which there is only a single copy, is essential for cell viability. Spinal muscular atrophy in humans results from homozygous mutations that inactivate SMN1. The small amount of protein translated from the small fraction of SMN2 mRNAs that are correctly spliced is sufficient to maintain cell viability during embryogenesis and fetal development, but it is not sufficient to maintain the viability of spinal cord motor neurons in childhood, resulting in their death and the associated disease.
Page 429
Approximately 15 percent of the single-base mutations that cause human genetic diseases interfere with proper exon definition. Some of these mutations occur in 5′ or 3′ splice sites, often resulting in the use of nearby alternative “cryptic” splice sites that are present in the normal gene sequence. In the absence of the normal splice site, the cross-exon recognition complex recognizes these alternative sites. Other mutations that cause abnormal splicing result in a new consensus splice-site sequence that becomes recognized in place of the normal splice site. Finally, some mutations can interfere with the binding of specific SR proteins to pre-mRNAs. These mutations inhibit splicing at normal splice sites, as in the case of the SMN2 gene, and thus lead to exon skipping.
Strategies involving membrane-permeant synthetic oligonucleotide derivatives similar to those discussed above for causing skipping of mutant exons in DMD are being developed for the treatment of these genetic diseases. Such molecules can hybridize to a mutant sequence that creates an abnormal splice site, sterically blocking access of U1 or U2 snRNAs to that site. In the case of spinal muscular atrophy, researchers are experimenting with modified oligonucleotides that base-pair to a region in the SMN2 pre-mRNA close to the missing exonic splicing enhancer. A non-hybridizing region that remains single-stranded and can bind an abundant SR protein may help to assemble a cross-exon recognition complex to increase correct splicing of exons in pre-mRNAs expressed from the SMN2 gene.