Mutations in Mitochondrial DNA Cause Several Genetic Diseases in Humans
The severity of disease caused by a mutation in mtDNA depends on the nature of the mutation and on the proportion of mutant and wild-type mtDNAs present in a particular cell type. Generally, when mutations in mtDNA are found, cells contain mixtures of wild-type and mutant mtDNAs—a condition known as heteroplasmy. Each time a mammalian somatic or germ-line cell divides, the mutant and wild-type mtDNAs segregate randomly into the daughter cells, as occurs in yeast cells (see Figure 12-9b). Thus the mtDNA genotype, which fluctuates from one generation and from one cell division to the next, can drift toward predominantly wild-type or predominantly mutant mtDNAs. Since all enzymes required for the replication and growth of mammalian mitochondria, such as the mitochondrial DNA and RNA polymerases, are encoded in the nucleus and imported from the cytosol, a mutant mtDNA should not be at a “replication disadvantage”; mutants that have large deletions of mtDNA might even be at a selective advantage because they can replicate faster.
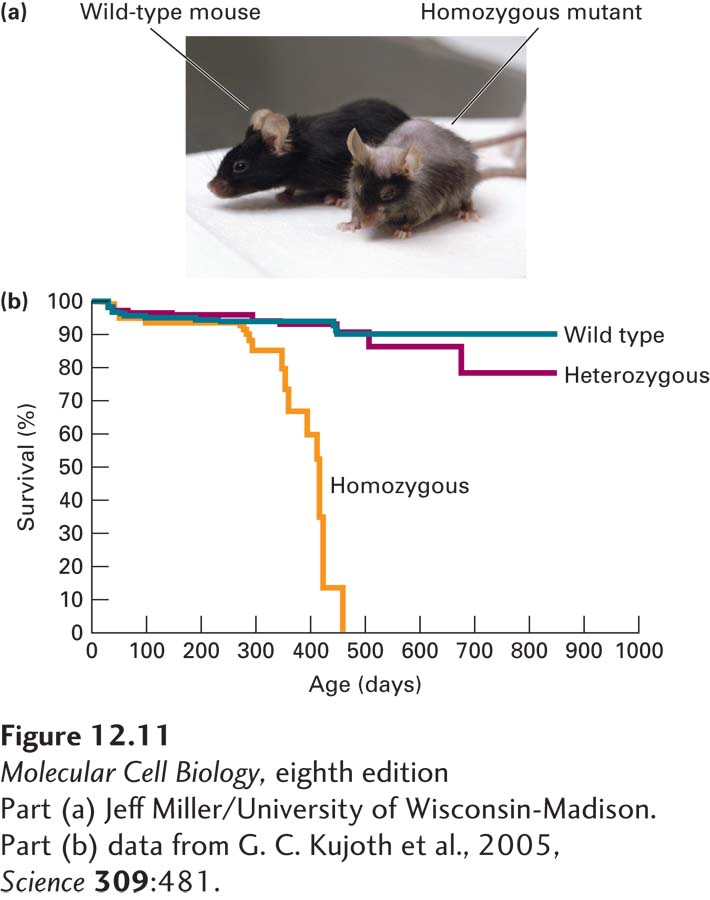
Recent research suggests that the accumulation of mutations in mtDNA is an important component of aging in mammals. Mutations in mtDNA have been observed to accumulate over time, probably because mammalian mtDNA is not repaired in response to DNA damage. To study this hypothesis, researchers used gene “knock-in” techniques in mice to replace the nuclear gene encoding mitochondrial DNA polymerase with normal proofreading activity (see Figure 5-33) with a mutant gene encoding a polymerase that is defective in proofreading. Mutations in mtDNA accumulated much more rapidly in homozygous mutant mice than in wild-type mice, and the mutant mice aged at a highly accelerated rate and died earlier than wild-type mice (Figure 12-11). It has been proposed that the loss of mitochondrial function that accompanies aging, due in part to accumulation of mutations and damage induced by reactive oxygen species, might contribute to aging and limit the life span. However, additional studies will be required to determine how mitochondrial dysfunction, aging, and longevity are related.
[Part (a) Jeff Miller/University of Wisconsin-Madison. Part (b) data from G. C. Kujoth et al., 2005, Science309:481.]
EXPERIMENTAL FIGURE 12-11Mice with a mitochondrial DNA polymerase defective for proofreading exhibit premature aging. A line of “knock-in” mice were prepared by methods discussed in Chapter 6 with an aspartic acid-to-alanine mutation in the gene encoding mitochondrial DNA polymerase (D257A), which inactivated the polymerase’s proofreading function. (a) Wild-type and homozygous mutant mice at 390 days old (13 months). The mutant mouse displays many of the features of an aged mouse (>720 days, or 24 months, of age). (b) Plot of survival versus time of wild-type (+/+), heterozygous (D257A/+), and homozygous (D257A/D257A) mice.
[Part (a) Jeff Miller/University of Wisconsin-Madison. Part (b) data from G. C. Kujoth et al., 2005, Science309:481.]
With few exceptions, all human cells have mitochondria, yet mutations in mtDNA affect only some tissues. Those most commonly affected are tissues that have a high requirement for the ATP produced by oxidative phosphorylation and tissues that require most or all of the mtDNA in the cell to synthesize sufficient amounts of functional mitochondrial proteins. For instance, Leber’s hereditary optic neuropathy (degeneration of the optic nerve) is caused by a missense mutation in the mtDNA gene encoding subunit 4 of the NADH-CoQ reductase (complex I), a protein required for ATP production by mitochondria (see below). Several large deletions in mtDNA cause another set of diseases, including chronic progressive external ophthalmoplegia, characterized by eye defects, and Kearns–Sayre syndrome, characterized by eye defects, an abnormal heartbeat, and central nervous system degeneration. A third condition, causing “ragged-red” muscle fibers (with improperly assembled mitochondria) and associated uncontrolled jerky movements, is due to a single mutation in the TΨCG loop of the mitochondrial lysine tRNA. As a result of this mutation, the translation of several mitochondrial proteins is apparently inhibited.