Improperly Folded Proteins in the ER Induce Expression of Protein-Folding Catalysts
Wild-type proteins that are synthesized on the rough ER cannot exit this compartment until they achieve their completely folded conformation. Likewise, almost any mutation that prevents proper folding of a protein in the ER also blocks the movement of that protein from the ER lumen or membrane to the Golgi complex. The mechanisms that retain unfolded or incompletely folded proteins within the ER probably increase the overall efficiency of folding by keeping intermediate forms in proximity to folding catalysts, which are most abundant in the ER. Improperly folded proteins retained within the ER are generally seen bound to the ER chaperones BiP and calnexin. Thus these luminal folding catalysts perform two related functions: assisting in the folding of normal proteins by preventing their aggregation and binding to misfolded proteins to retain them in the ER.
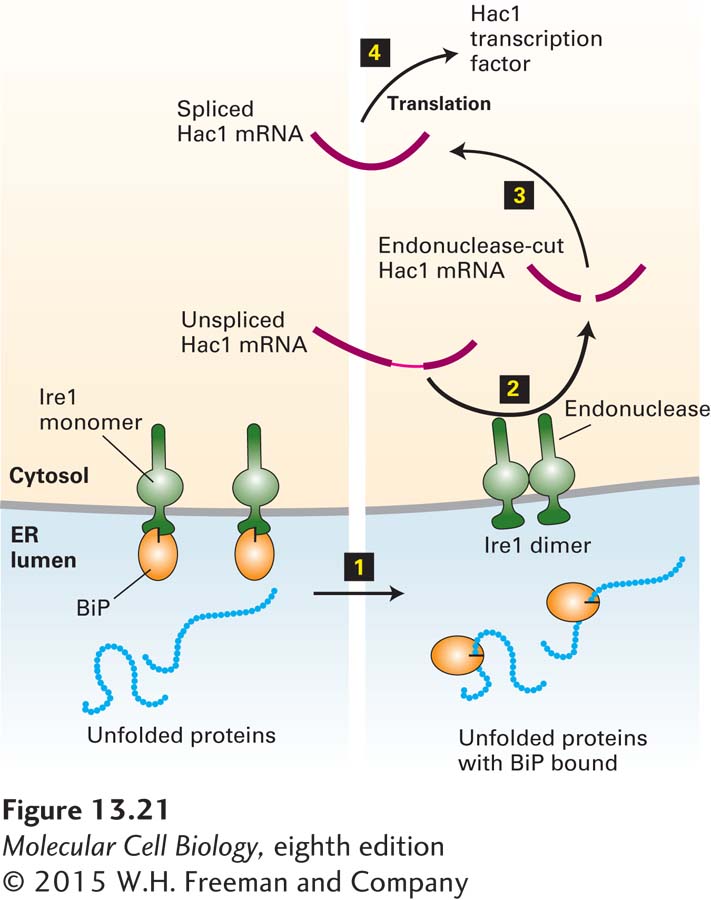
Both mammalian cells and yeasts respond to the presence of unfolded proteins in the rough ER by increasing transcription of several genes that encode ER chaperones and other folding catalysts. A key participant in this unfolded-protein response is Ire1, an ER membrane protein that exists both as a monomer and as a dimer. The dimeric form, but not the monomeric form, promotes formation of Hac1, a transcription factor in yeast that activates expression of the genes induced in the unfolded-protein response. As depicted in Figure 13-21, binding of BiP to the luminal domain of monomeric Ire1 prevents formation of the Ire1 dimer. Thus the quantity of free BiP in the ER lumen determines the relative proportions of monomeric and dimeric Ire1. Accumulation of unfolded proteins within the ER lumen sequesters BiP molecules, making them unavailable for binding to Ire1. As a result, the level of dimeric Ire1 increases, leading to an increase in the level of Hac1 and production of proteins that assist in protein folding.
FIGURE 13-21The unfolded-protein response. Ire1, a transmembrane protein in the ER membrane, has a binding site for BiP on its luminal domain; the cytosolic domain contains a specific RNA endonuclease. Step 1: Accumulating unfolded proteins in the ER lumen bind BiP molecules, releasing them from monomeric Ire1. Dimerization of Ire1 then activates its endonuclease activity. Steps 2–3: The unspliced mRNA precursor encoding the transcription factor Hac1 is cleaved by dimeric Ire1, and the two exons are joined to form functional Hac1 mRNA. Current evidence indicates that this processing occurs in the cytosol, although pre-mRNA processing generally occurs in the nucleus. Step 4: Hac1 is translated into Hac1 protein, which then moves back into the nucleus and activates transcription of genes encoding several protein-folding catalysts. See U. Ruegsegger et al., 2001, Cell107:103; A. Bertolotti et al., 2000, Nat. Cell Biol.2:326; and C. Sidrauski and P. Walter, 1997, Cell90:1031.
Page 607
Mammalian cells contain an additional regulatory pathway that operates in response to unfolded proteins in the ER. In this pathway, accumulation of unfolded proteins in the ER triggers proteolysis of ATF6, a transmembrane protein in the ER membrane, at a site within the membrane-spanning segment. The cytosolic domain of ATF6 released by proteolysis then moves to the nucleus, where it stimulates transcription of the genes encoding ER chaperones. Activation of a transcription factor by such regulated intramembrane proteolysis also occurs in the Notch signaling pathway and during activation of the cholesterol-responsive transcription factor SREBP (see Figures 16-36 and 16-38).
A hereditary form of emphysema illustrates the detrimental effects that can result from misfolding of proteins in the ER. This disease is caused by a point mutation in α1-antitrypsin, which is normally secreted by hepatocytes and macrophages. The wild-type protein binds to and inhibits trypsin as well as the blood protease elastase. In the absence of normal α1-antitrypsin, elastase degrades the fine tissue in the lung that participates in the absorption of oxygen, eventually producing the symptoms of emphysema. Although the mutant α1-antitrypsin is synthesized on the rough ER, it does not fold properly, forming an almost crystalline aggregate that is not exported from the ER. In hepatocytes, the secretion of other proteins also becomes impaired as the rough ER is filled with aggregated α1-antitrypsin.