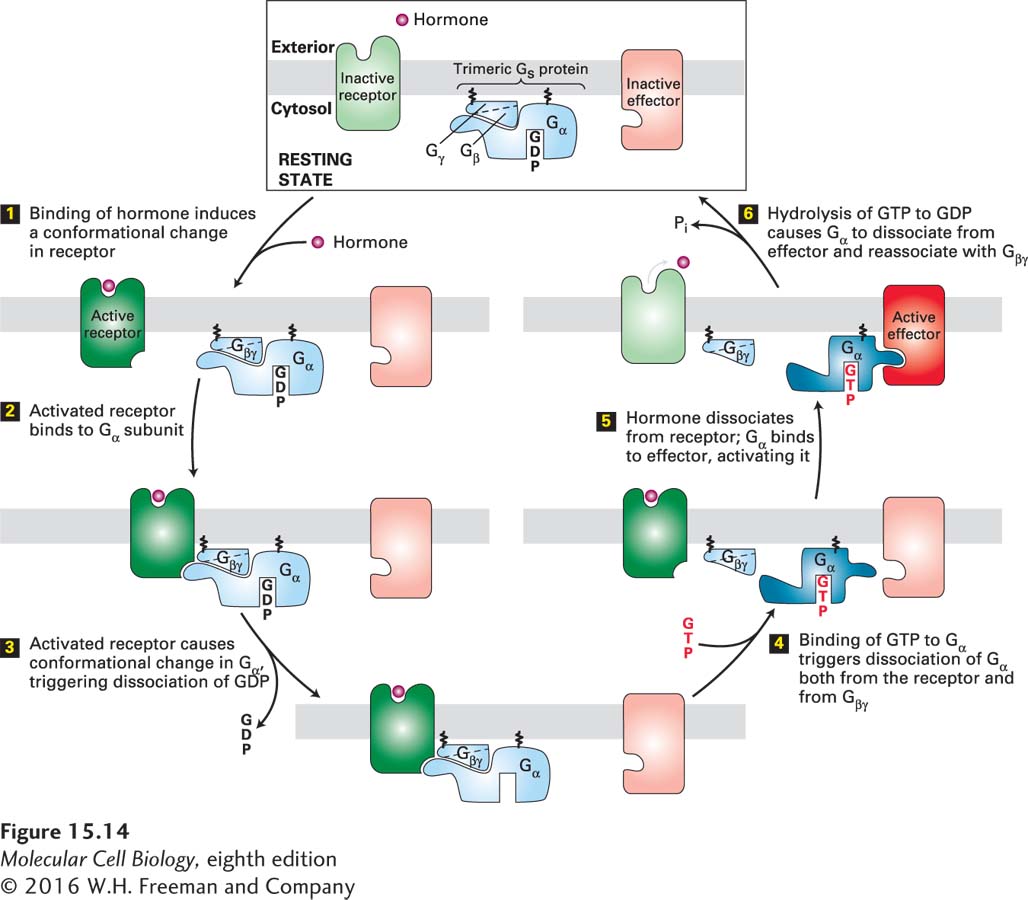
Ligand-Activated G Protein–Coupled Receptors Catalyze Exchange of GTP for GDP on the α Subunit of a Heterotrimeric G Protein
Heterotrimeric G proteins contain three subunits, designated α, β, and γ. Only the Gα subunit binds GTP or GDP; both the Gα and Gγ subunits are linked to the plasma membrane by covalently attached lipids. The β and γ subunits are always bound together and are usually referred to as the Gβγ subunit. In the resting state, when no ligand is bound to the receptor, the Gα subunit has a bound GDP and is complexed with Gβγ. Binding of a ligand (e.g., epinephrine) or an agonist (e.g., isoproterenol) to a G protein–coupled receptor changes the conformation of its transmembrane helices and enables the receptor to bind to the Gα subunit of the intact heterotrimeric G protein (Figure 15-14, steps 1 and 2). This binding releases the bound GDP; thus the activated ligand-bound receptor functions as a guanine nucleotide exchange factor (GEF) for the Gα subunit (step 3). Next GTP rapidly binds to the “empty” guanine nucleotide site in the Gα subunit, causing a change in the conformation of its switch segments (see Figure 15-5). These changes weaken the binding of Gα to both the receptor and the Gβγ subunit (step 4). In most cases, Gα·GTP, which remains anchored in the plasma membrane, then interacts with and activates an effector protein (step 5). In some cases, Gα·GTP inhibits, rather than activates, the effector. Moreover, depending on the type of cell and G protein involved, the Gβγ subunit, freed from the Gα subunit, will sometimes transduce a signal by interacting with an effector protein.
FIGURE 15-14General mechanism of the activation of effector proteins associated with G protein–coupled receptors. Light colors denote the inactive and dark colors the active conformations of each protein. The Gα and Gβγ subunits of a heterotrimeric G protein are tethered to the membrane by covalently attached lipid molecules (wiggly black lines). Following ligand binding, exchange of GDP for GTP, and dissociation of the G protein subunits (steps 1 – 4), the free Gα·GTP binds to and activates an effector protein (step 5). Hydrolysis of GTP terminates signaling and leads to reassembly of the heterotrimeric G protein, returning the system to the resting state (step 6). Binding of another ligand molecule causes repetition of the cycle. In some pathways, the effector protein is activated by the free Gβγ subunit. See W. Oldham and H. Hamm, 2006, Quart. Rev. Biophys. 39:117.
The active Gα·GTP state is relatively short-lived because hydrolysis of the bound GTP to GDP, catalyzed by the intrinsic GTPase activity of the Gα subunit, occurs in minutes (Figure 15-14, step 6). The conformation of the Gα subunit thus switches back to the inactive Gα·GDP state, blocking any further activation of effector proteins. The resulting Gα·GDP quickly reassociates with Gβγ, and the complex becomes ready to interact with an activated receptor and start the process all over again.
Page 690
The rate of GTP hydrolysis is sometimes further enhanced by binding of the Gα·GTP complex to the effector; in this case, the effector functions as a GTPase-activating protein (GAP). This feedback mechanism significantly reduces the duration of effector activation and avoids a cellular overreaction. In many cases, a second type of GAP protein, called a regulator of G protein signaling (RGS), also accelerates GTP hydrolysis by the Gα subunit, further reducing the time during which the effector remains activated. Thus the GPCR signal transduction system contains built-in feedback mechanisms that ensure that the effector protein becomes activated for only a few seconds or minutes following receptor activation; continual activation of receptors via ligand binding, together with subsequent activation of the corresponding G protein, is essential for prolonged activation of the effector.
Early evidence supporting the model shown in Figure 15-14 came from studies with compounds that are structurally similar to GTP, and so can bind to Gα subunits as well as GTP does, but cannot be hydrolyzed by the intrinsic GTPase. In some of these compounds, the P–O–P phosphodiester linkage connecting the β and γ phosphates of GTP is replaced by a nonhydrolyzable P–CH2–P or P–NH–P linkage. Addition of such a GTP analog to a plasma-membrane preparation in the presence of an agonist for a particular receptor results in a much longer-lived activation of the Gα protein and its associated effector protein than occurs with GTP. In this experiment, once the nonhydrolyzable GTP analog is exchanged for GDP bound to Gα, it remains permanently bound to Gα. Because the Gα·GTP-analog complex is as functional as the normal Gα·GTP complex in activating the effector protein, the effector remains permanently active.
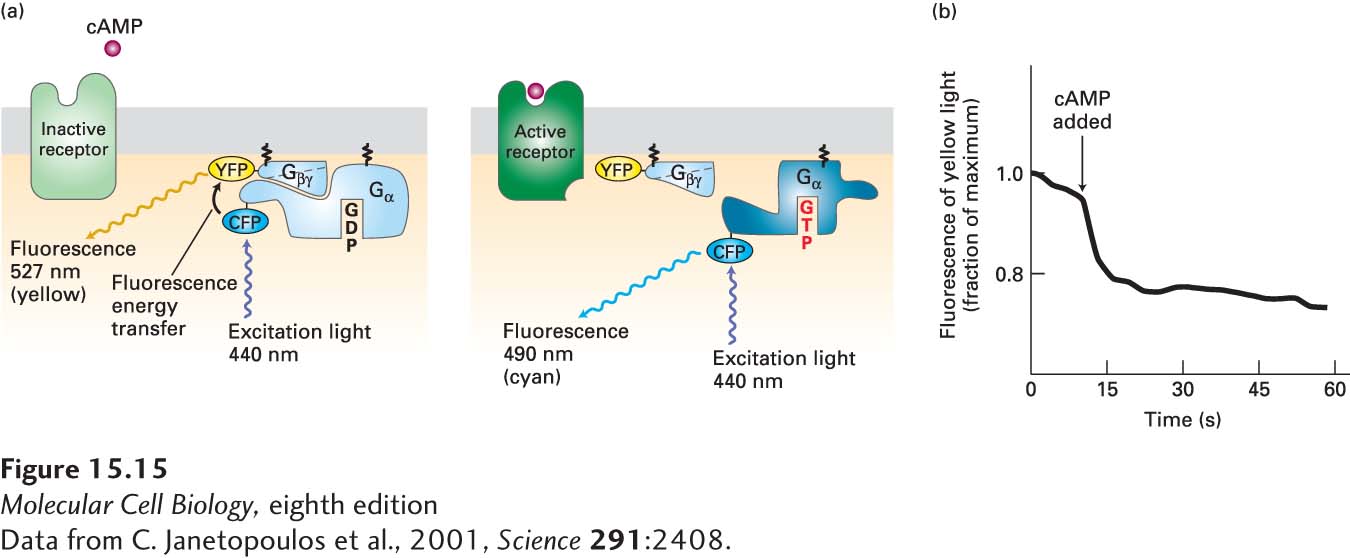
GPCR-mediated dissociation of heterotrimeric G proteins can be detected in live cells. These studies have exploited the phenomenon of Förster resonance energy transfer (FRET), which changes the wavelength of emitted fluorescence when two fluorescent proteins interact (see Figure 4-24). Figure 15-15 shows how this experimental approach has demonstrated the dissociation of the Gα·Gβγ complex within a few seconds of ligand addition, providing evidence for the model of G protein cycling. This general experimental approach can be used to follow the formation and dissociation of other protein-protein complexes in live cells.
[Data from C. Janetopoulos et al., 2001, Science291:2408.]
EXPERIMENTAL FIGURE 15-15Activation of a G protein occurs within seconds of ligand binding to its cell-surface G protein–coupled receptor. In the amoeba Dictyostelium discoideum, cAMP acts as an extracellular signaling molecule and binds to and signals via a G protein–coupled receptor; it is not a second messenger. Amoeba cells were transfected with genes encoding two fusion proteins: a Gα fused to cyan fluorescent protein (CFP), and a Gβ fused to yellow fluorescent protein (YFP). CFP normally fluoresces 490-nm light; YFP, 527-nm light. (a) When CFP and YFP are close to each other, as in the resting Gα·Gβγ complex, fluorescence energy transfer can occur between them (left). As a result, irradiation of resting cells with 440-nm light (which directly excites CFP but not YFP) causes emission of 527-nm (yellow) light, characteristic of YFP, because of fluorescence energy transfer from CFP to YFP. However, if ligand binding leads to dissociation of the Gα and Gβγ subunits, then fluorescence energy transfer cannot occur. In this case, irradiation of cells at 440 nm causes emission of 490-nm (cyan) light, characteristic of CFP (right). (b) Plot of the emission of yellow light (527 nm) from a single transfected amoeba cell before and after addition of extracellular cAMP (arrow). The drop in yellow fluorescence, which results from the dissociation of the Gα-CFP fusion protein from the Gβ-YFP fusion protein, occurs within seconds of cAMP addition.
[Data from C. Janetopoulos et al., 2001, Science291:2408.]
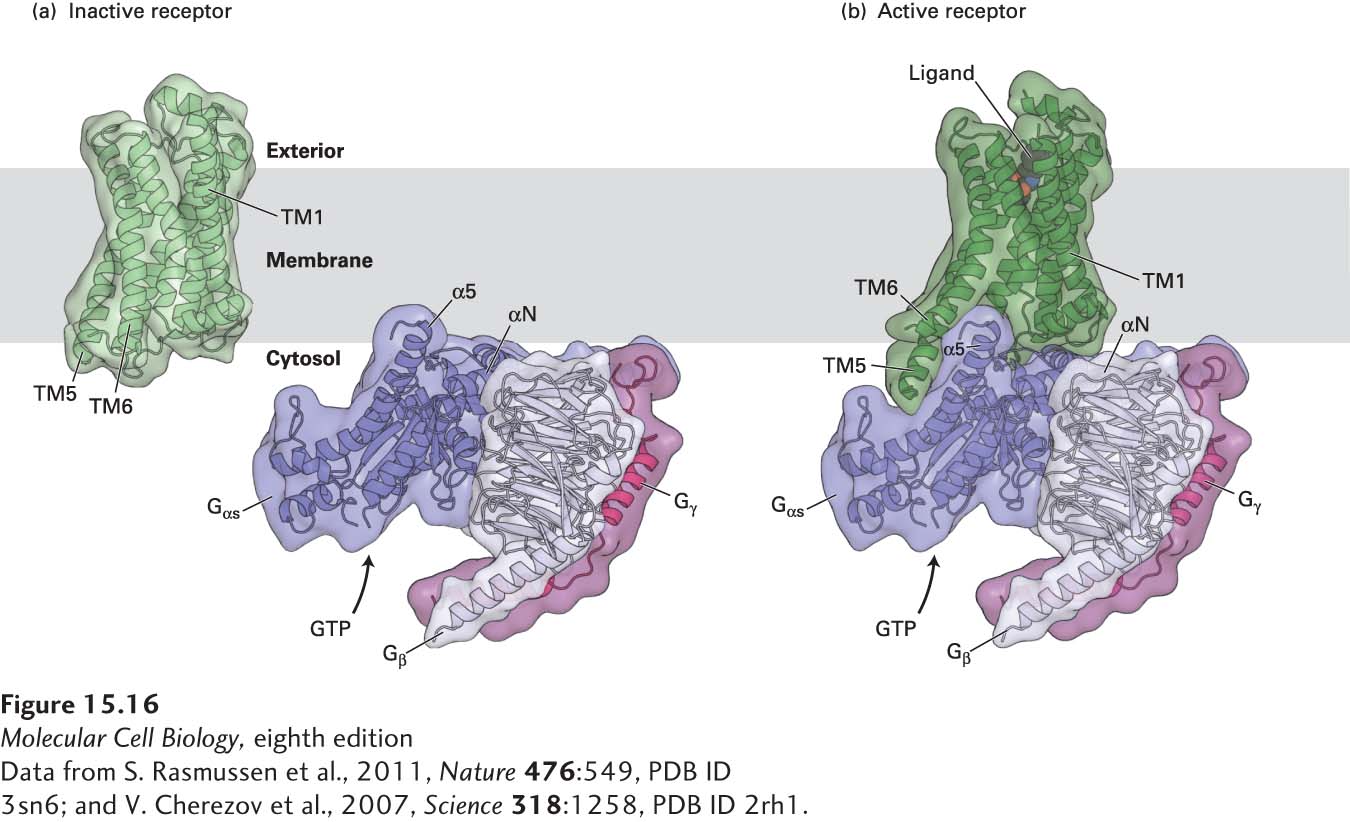
For many years, it was impossible to determine the structure of the same GPCR in the active and inactive states. This has now been accomplished with the β2-adrenergic receptor (as well as with rhodopsin, discussed in Section 15.4). The initial cloning and characterization of the β2-adrenergic receptor by Robert Lefkowitz and the three-dimensional structures obtained by Brian Kobilka and depicted in Figure 15-16 were rewarded with the 2012 Nobel Prize in Chemistry. The seven membrane-embedded α helices of the β2-adrenergic receptor form the binding pocket to which an agonist or antagonist can be noncovalently bound (see Figure 15-13). Binding of an agonist to the receptor induces major conformational changes in which there are substantial movements of transmembrane helices 5 and 6 and changes in the structure of the C3 loop; together, these changes create a surface that can now bind to a segment of the Gα subunit (Figure 15-16b). Note that when unbound by ligand, the receptor (Figure 15-16a) has no surface that is complementary to the G protein and thus cannot bind to it.
[Data from S. Rasmussen et al., 2011, Nature476:549, PDB ID 3sn6; and V. Cherezov et al., 2007, Science318:1258, PDB ID 2rh1.]
FIGURE 15-16Structure of theβ2-adrenergic receptor in the inactive and active states and with its associated heterotrimeric G protein, Gs. (a) The three-dimensional structure of the β2-adrenergic receptor bound to an antagonist (not shown) and thus in the inactive state. Placed next to it are the three-dimensional structures of the subunits of the heterotrimeric G protein Gs: Gαs (dark purple), Gβ (light purple), and Gγ (pink), showing the inability of the resting receptor to bind to and activate Gαs. (b) The overall structure of the active receptor complex shows the adrenergic receptor bound to an agonist (black and red spheres) and engaged in extensive interactions with a segment of Gαs.
[Data from S. Rasmussen et al., 2011, Nature476:549, PDB ID 3sn6; and V. Cherezov et al., 2007, Science318:1258, PDB ID 2rh1.]
Page 691
X-ray crystallographic studies of the complex of the activated β2-adrenergic receptor and its coupled G protein, Gs, have also revealed how the subunits of a G protein interact with each other and have provided clues about how binding of GTP leads to dissociation of the Gα from the Gβγ subunit. Upon binding to an activated receptor, the Gα subunit undergoes a small conformational change, lengthening the α5 helix. This change creates a large surface, mainly consisting of the N-terminal α-helical segments αN and α5 of the Gαs protein that bind mainly to transmembrane helices 5 and 6 of the activated receptor (see Figure 15-16b). Concomitantly, this conformational change in Gα triggers the release of GDP. Note that a large surface of Gα·GDP, including αN, interacts with the Gβ subunit, but Gα·GDP does not directly contact Gγ. Binding to the GPCR is followed by opening of the Gα subunit, eviction of the bound GDP, and its replacement with GTP; this is immediately followed by conformational changes within switches I and II that disrupt the molecular interactions between Gα and Gβγ, leading to the dissociation of Gα from the Gβγ subunit.