Degradation of an Inhibitor Protein Activates the NF-κB Transcription Factor
In the resting state of both the Wnt and Hedgehog pathways, a key transcription factor is ubiquitinylated and subjected to proteolytic degradation, generating a protein fragment that acts as a transcriptional repressor; activation of the signaling pathway involves blockage of ubiquitinylation and release of the transcription factor in its active state. The NF-κB pathway works in the opposite manner: in the resting state, the NF-κB transcription factor is retained in the cytosol bound to an inhibitor; activation of the signaling pathway involves ubiquitinylation of the inhibitor followed by its degradation, triggering release of the active transcription factor. This mechanism allows cells to respond to a variety of stress signals by immediately and vigorously activating gene transcription. The steps in the NF-κB pathway were revealed in studies with both mammalian cells and Drosophila.
Page 758
NF-κB (an acronym for the somewhat unwieldy descriptor “nuclear-factor kappa-light-chain enhancer of activated B cells”) is rapidly activated in mammalian immune-system cells in response to bacterial and viral infection, inflammation, and a number of other stressful situations, such as ionizing radiation. The NF-κB pathway is activated in some cells of the immune system when components of bacterial or fungal cell walls bind to certain Toll-like receptors on the cell surface (see Figure 23-35). This pathway is also activated by so-called inflammatory cytokines, such as tumor necrosis factor alpha (TNFα) and interleukin 1 (IL-1), which are released by nearby cells in response to infection. In all of these cases, binding of ligand to its receptor induces assembly of a multiprotein complex in the cytosol near the plasma membrane that triggers a signaling pathway resulting in activation of the NF-κB transcription factor.
NF-κB was originally discovered on the basis of its transcriptional activation of the gene encoding the light chains of antibodies (immunoglobulins) in B cells. It is now thought to be the master transcriptional regulator of the immune system in mammals. Although flies do not make antibodies, NF-κB homologs in Drosophila induce synthesis of a large number of secreted antimicrobial peptides in response to bacterial and viral infection. This phenomenon indicates that the NF-κB regulatory system has been conserved during evolution and is more than half a billion years old.
Page 759
Page 760
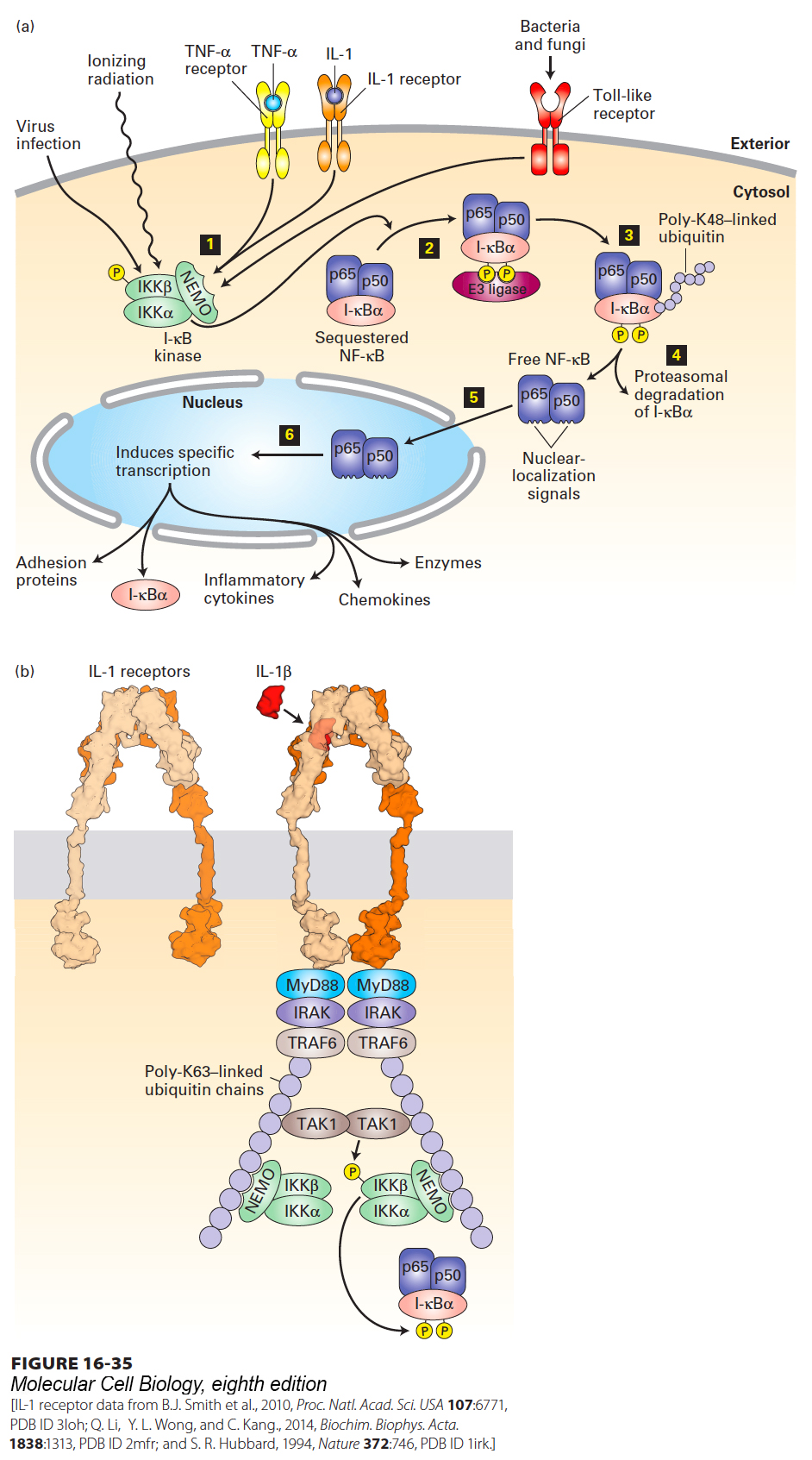
Biochemical studies in mammalian cells and genetic studies in flies have provided important insights into the operation of the NF-κB pathway. The two subunits (p65 and p50) of the heterodimeric NF-κB transcription factor share a region of homology at their N-termini that is required for their dimerization and binding to DNA. In cells that are not undergoing a stress or responding to signs of an infection, direct binding to an inhibitor called I-κBα sequesters NF-κB in an inactive state in the cytosol. A single molecule of I-κBα binds to the paired N-terminal domains of the p50–p65 subunits, thereby masking their nuclear-localization signals (Figure 16-35a).
[IL-1 receptor data from B.J. Smith et al., 2010, Proc. Natl. Acad. Sci. USA107:6771, PDB ID 3loh; Q. Li, Y. L. Wong, and C. Kang., 2014, Biochim. Biophys. Acta. 1838:1313, PDB ID 2mfr; and S. R. Hubbard, 1994, Nature372:746, PDB ID 1irk.]
FIGURE 16-35Activation of the NF-κB signaling pathway. (a) In resting cells, the dimeric transcription factor NF-κB, composed of p50 and p65 subunits, is sequestered in the cytosol, bound to the inhibitor I-κBα. Step 1: Activation of the trimeric I-κB kinase is stimulated by many agents, including viral infection, ionizing radiation, binding of the pro-inflammatory cytokines TNFα or IL-1 to their respective receptors, or activation of any of several Toll-like receptors by components of invading bacteria or fungi. Step 2: The β subunit of I-κB kinase then phosphorylates the inhibitor I-κBα, which then binds an E3 ubiquitin ligase. Steps 3 and 4: Subsequent lysine 48–linked polyubiquitinylation of I-κBα targets it for degradation by proteasomes. Step 5: The removal of I-κBα unmasks the nuclear-localization signals in both subunits of NF-κB, allowing their translocation to the nucleus. Step 6: In the nucleus, NF-κB activates transcription of numerous target genes, including the gene encoding I-κBα, which acts to terminate signaling, and genes encoding various inflammatory cytokines. See R. Khush et al., 2001, Trends Immunol.22:260, and J-L Luo et al., 2005, J. Clin. Invest. 115:2625. (b) Binding of interleukin-1β (IL-1β) to heterodimeric IL-1 receptors triggers receptor oligomerization and recruitment of several proteins to the receptor cytosolic domain, including TRAF6, an E3 ubiquitin ligase, which catalyzes synthesis of long lysine-63-linked polyubiquitin chains linked to TRAF6, NEMO, and other proteins in the complex. The polyubiquitin chains function as a scaffold to recruit the kinase TAK1 and the NEMO subunit of the trimeric I-κB kinase complex. TAK1 then phosphorylates itself and the β subunit of I-κB kinase, activating its kinase activity and enabling it to phosphorylate I-κBα. See B. Skaug et al., 2009, Annu. Rev. Biochem.78:769, and J. Napetschnig and H. Wu, 2013 Annu. Rev. Biophys.42:443.
[IL-1 receptor data from B.J. Smith et al., 2010, Proc. Natl. Acad. Sci. USA107:6771, PDB ID 3loh; Q. Li, Y. L. Wong, and C. Kang., 2014, Biochim. Biophys. Acta. 1838:1313, PDB ID 2mfr; and S. R. Hubbard, 1994, Nature372:746, PDB ID 1irk.]
A three-protein complex termed I-κB kinase operates immediately upstream of NF-κB and is responsible for releasing it from sequestration. The β subunit of I-κB kinase is the point of convergence of all of the extracellular signals noted above that activate NF-κB. Within minutes of stimulation of the cell by an infectious agent or inflammatory cytokine, the β subunit of I-κB kinase becomes activated by phosphorylation and then phosphorylates two N-terminal serine residues on I-κBα (Figure 16-35a, steps 1 and 2). An E3 ubiquitin ligase then binds to these phosphoserines and polyubiquitinylates I-κBα, triggering its immediate degradation by a proteasome (steps 3 and 4). In cells expressing mutant forms of I-κBα in which these two serines have been changed to alanine and so cannot be phosphorylated, NF-κB is permanently inactive, demonstrating that phosphorylation of I-κBα is essential for pathway activation.
The degradation of I-κBα exposes the nuclear-localization signals on NF-κB, which then translocates into the nucleus and activates transcription of a multitude of target genes (Figure 16-35a, steps 5 and 6). Despite its activation by proteolysis, NF-κB signaling is eventually turned off by a negative feedback loop because one of the genes whose transcription is immediately induced by NF-κB encodes I-κBα. The resulting increased levels of the I-κBα protein bind active NF-κB in the nucleus and return it to the cytosol.
In many immune-system cells, NF-κB stimulates transcription of more than 150 genes, including those encoding cytokines and chemokines; the latter attract other immune-system cells and fibroblasts to sites of infection. NF-κB also promotes expression of receptor proteins that enable neutrophils (a type of white blood cell) to migrate from the blood into the underlying tissue (see Figure 20-40). In addition, NF-κB stimulates expression of iNOS, the inducible isoform of the enzyme that produces nitric oxide (see Figure 15-36), which is toxic to bacterial cells, as well as expression of several anti-apoptotic proteins, which prevent cell death. Thus this single transcription factor coordinates and activates the body’s defense, either directly by responding to pathogens and stress or indirectly by responding to signaling molecules released from other infected or wounded tissues and cells.