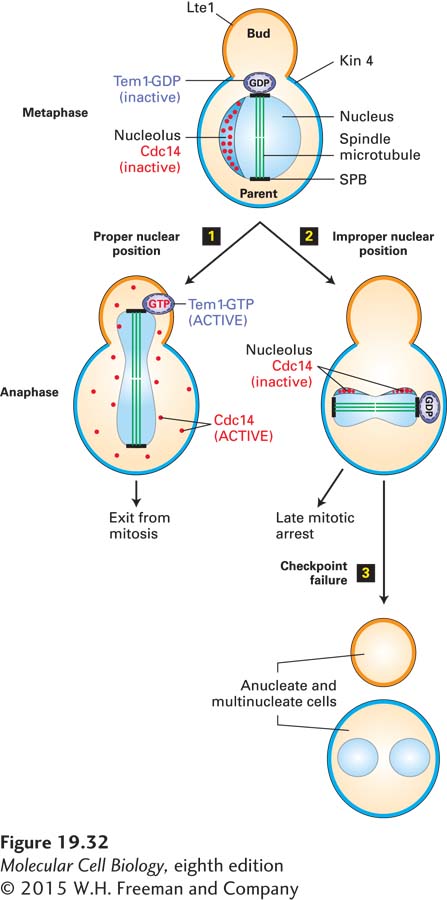
The Spindle Position Checkpoint Pathway Ensures That the Nucleus Is Accurately Partitioned Between Two Daughter Cells
The coordination of the site of nuclear division with that of cytokinesis is essential for the production of two identical daughter cells. If cytokinesis occurred in such a way that each daughter cell failed to receive a complete genetic complement, chromosome loss or gain would ensue. A surveillance mechanism that prevents cytokinesis when the mitotic spindle is not correctly positioned in the cell has been described in many model systems. This surveillance mechanism, known as the spindle position checkpoint pathway, is best understood in budding yeast. In this organism, the site of bud formation, and therefore the site of cytokinesis, is determined during G1. Thus the axis of division is defined prior to mitosis, and the mitotic spindle must be aligned along this parent-bud axis during every cell division (Figure 19-32, step 1). When this process fails, the spindle position checkpoint prevents mitotic CDK inactivation, giving the cell an opportunity to reposition the spindle prior to spindle disassembly and cytokinesis (Figure 19-32, step 2). If the spindle position checkpoint fails, cells that misposition their spindles give rise to mitotic products with too many or too few nuclei (Figure 19-32, step 3).
FIGURE 19-32The spindle position checkpoint pathway in budding yeast. Cdc14 phosphatase activity is required for exit from mitosis. Top: In S. cerevisiae, during interphase and early mitosis, Cdc14 (red dots) is sequestered and inactivated in the nucleolus. Inactive Tem1-GDP (purple) associates with the spindle pole body (SPB) nearest to the bud as soon as the mitotic spindle forms. If chromosome segregation occurs properly step 1, extension of the spindle microtubules inserts the daughter SPB into the bud, causing Tem1 to be activated by an unknown mechanism. Tem1-GTP activates a protein kinase cascade, which then promotes the release of active Cdc14 from the nucleolus and exit from mitosis. If the spindle apparatus fails to place the daughter SPB in the bud step 2, Kin4 (cyan), an inhibitor of Tem1, is recruited from the parent cell cortex to the parent-cell-located SPB and maintains Tem1 in the GDP-bound form, and mitotic exit does not occur. Lte1 (orange) is an inhibitor of Kin4 and is localized to the bud. Lte1 prevents Kin4 that leaks into the bud from inhibiting Tem1. If the checkpoint fails step 3, cells with mispositioned spindles inappropriately exit mitosis and produce anucleate and multinucleate cells.
Page 910
Recall that in budding yeast, a pool of mitotic cyclins is spared from degradation by APC/CCdc20 to facilitate the sometimes difficult process of aligning the mitotic spindle in such a way that half the nucleus squeezes through the tiny bud neck during anaphase spindle elongation. Recall further that inactivation of this protected pool of mitotic cyclin-CDK complexes is triggered by the protein phosphatase Cdc14, which in turn is activated by a signal transduction pathway known as the mitotic exit network (see Figure 19-26). The mitotic exit network is controlled by a small (monomeric) GTPase called Tem1. This member of the GTPase superfamily of switch proteins controls the activity of a protein kinase cascade in a manner similar to the way Ras controls MAP kinase pathways (see Chapter 16). Tem1 associates with spindle pole bodies as soon as they form. An inhibitor of the GTPase, called Kin4, localizes to the parent cell but is absent from the bud (see Figure 19-32). An inhibitor of Kin4, called Lte1, localizes to the bud but is absent from the parent cell; it inhibits any residual Kin4 that leaks into the bud. When spindle microtubule elongation at the end of anaphase has correctly positioned segregating daughter chromosomes in the bud, Tem1 is released from inhibition by Kin4. As a consequence, Tem1 is converted into its active GTP-bound state, activating the protein kinase signaling cascade. The terminal kinase in the cascade then phosphorylates the nucleolar anchor that binds and inhibits Cdc14, releasing Cdc14 phosphatase into the cytoplasm and nucleoplasm in both the bud and the parent cell (see Figure 19-32, step 1). Once active Cdc14 is available, mitotic CDKs are inactivated and cells exit mitosis. When the spindle fails to be positioned correctly, the Tem1-bearing spindle pole body fails to enter the bud, the mitotic exit network cannot be activated, and the cell is arrested in anaphase. Thus spatial restriction of the inhibitors and activators of a signal transduction pathway allows the cell to sense a spatial cue, spindle position, and translate it into regulation of a signal transduction pathway.
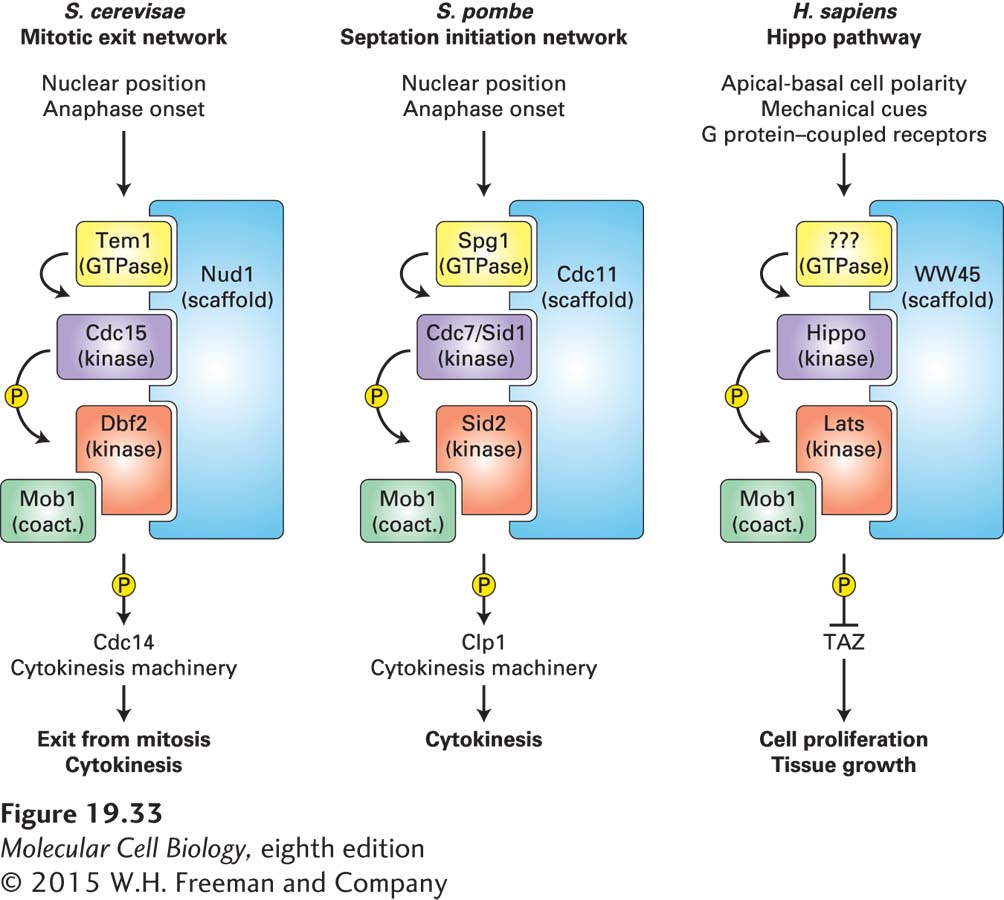
The mitotic exit network that localizes to spindle pole bodies and whose activity is regulated by spindle position belongs to the family of signaling pathways known as the Hippo pathway in metazoans and the septation initiation network in fission yeast (Figure 19-33). This pathway consists of a highly conserved core kinase signaling network, but its signaling input and output have diverged during evolution. The conserved core of the Hippo signaling pathways consists of the Hippo protein kinase, which activates the Lats-Mob1 kinase. The kinases are organized by a scaffolding molecule that, at least in budding and fission yeasts, targets the kinase cascade to centrosomes. As in many kinase signaling cascades, a GTPase controls the activity of the mitotic exit network and the septation initiation network. Whether the Hippo pathway is controlled by a GTPase is not yet known.
FIGURE 19-33The Hippo signaling pathway family. The core kinase signaling module of the Hippo pathway is conserved across species (homologous proteins are shown in the same color), but input signals as well as pathway effectors have diverged during evolution. In budding and fission yeasts, cell cycle signals such as anaphase onset and nuclear position regulate exit from mitosis and cytokinesis through effector molecules such as the protein phosphatase Cdc14 (Clp1 in fission yeast). In metazoans, tissue organization cues, such as pathways that establish cell polarity, mechanical cues, and signals from G protein–coupled receptors, activate the Hippo pathway. The Hippo pathway then inhibits the transcription factor TAZ to prevent cell proliferation and tissue growth.
Page 911
The Hippo pathway is a great example of how signaling pathways have been repurposed during evolution. In budding and fission yeasts, the pathway regulates CDK activity and cytokinesis in response to anaphase and nuclear position signals. In metazoans, the Hippo pathway restrains cell proliferation during G1. Inactivation of the Hippo pathway leads to tissue overgrowth because in the absence of Hippo pathway function, the transcriptional activator TAZ (known as YAP in the mouse) constitutively promotes the expression of growth- and proliferation-promoting genes. The signals controlling Hippo pathway function appear diverse. Apical-basal cell polarity via cell-junction components, G protein–coupled receptors, and mechanical forces activate the Hippo pathway and hence cause G1 arrest. Apical-basal polarity is characteristic of differentiated epithelial sheets. The exact mechanism whereby these polarity cues regulate the Hippo pathway is an area of active investigation, and the actin cytoskeleton appears to be a central node in this control. Cell junctions, mechanical forces, and G protein–coupled receptors all control the actin cytoskeleton and their regulators, such as the rho GTPases.
Given its central role in proliferation control, it is not surprising that the Hippo pathway plays a key role in stem cell maintenance and tissue regeneration. In fact, the pathway was first discovered because mutations in the Hippo pathway lead to tissue overgrowth and increased organ size in Drosophila. Its central function in restraining growth also means that the pathway is critically important for preventing tumor development. Mice harboring mutations in the Hippo pathway are cancer prone, and elevated levels of TAZ are frequently observed in human cancers.
This concludes our discussion of the surveillance mechanisms that ensure that cell cycle progression moves forward and occurs without error. As we will see in Chapter 24, cancer is a disease of failed cell cycle surveillance. Mutations in the pathways that ensure accurate chromosome replication, segregation, and nuclear partitioning are a major cause of cancerous transformation.