Type I and II Collagens Associate with Nonfibrillar Collagens to Form Diverse Structures
Collagens differ in the structures of the fibers they form and in how these fibers are organized into networks. Of the predominant types of collagen found in connective tissues, type I collagen forms long fibers, whereas networks of type II collagen are more mesh-like. In tendons, for instance, the long type I collagen fibers connect muscles to bones and must withstand enormous forces. Because type I collagen fibers have great tensile strength, tendons usually can be stretched without being broken. Indeed, gram for gram, type I collagen is stronger than steel. Two quantitatively minor fibrillar collagens, type V and type XI, co-assemble into fibers with type I collagen, thereby regulating the structures and properties of the fibers. Incorporation of type V collagen, for example, results in smaller-diameter fibers.
Page 953
Type I collagen fibrils are also used as the reinforcing rods in the construction of bone. Bones and teeth are hard and strong because they contain large amounts of dahllite, a crystalline calcium- and phosphate-containing mineral. Most bones are about 70 percent mineral and 30 percent protein, the vast majority of which is type I collagen. Bones form when certain cells (chondrocytes and osteoblasts) secrete collagen fibrils that are then mineralized by deposition of small dahllite crystals.
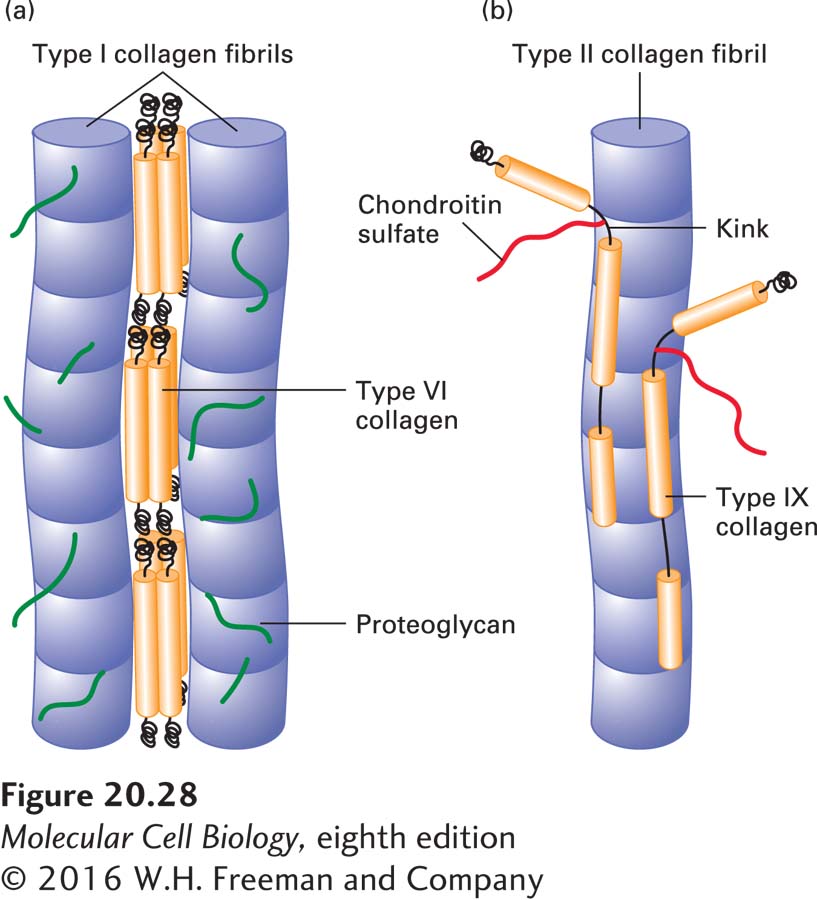
In many connective tissues, particularly skeletal muscle, proteoglycans and a fibril-associated collagen called type VI collagen are noncovalently bound to the sides of type I fibrils and may bind the fibrils together to form thicker collagen fibers (Figure 20-28a). Type VI collagen is unusual in that the molecule consists of a relatively short triple helix with globular domains at both ends. The lateral association of two type VI monomers generates an “antiparallel” dimer. The end-to-end association of these dimers through their globular domains forms type VI “microfibrils.” These microfibrils have a beads-on-a-string appearance, with about 60-nm-long triple-helical regions separated by 40-nm-long globular domains.
FIGURE 20-28Interactions of fibrillar collagens with fibril-associated collagens. (a) In tendons, type I fibrils are all oriented in the direction of the stress applied to the tendon. Proteoglycans and type VI collagen bind noncovalently to type I fibrils, coating the surface. The microfibrils of type VI collagen, which contain globular and triple-helical segments, bind to type I fibrils and link them together into thicker fibers. See R. R. Bruns et al., 1986, J. Cell Biol.103:393. (b) In cartilage, type IX collagen molecules are covalently bound at regular intervals along type II fibrils. A chondroitin sulfate chain, covalently linked to the α2(IX) chain at the flexible kink, projects outward from the fibril, as does the globular N-terminal region. See L. M. Shaw and B. Olson, 1991, Trends Biochem. Sci.18:191
The fibrils of type II collagen, the major collagen in cartilage, are smaller in diameter than type I fibrils and are oriented randomly in a viscous proteoglycan matrix. The rigid collagen fibrils impart strength to the matrix and allow it to resist large deformations. Type II fibrils are cross-linked to matrix proteoglycans by type IX collagen, another fibril-associated collagen. Type IX collagen and several related types have two or three triple-helical segments connected by flexible kinks and a globular N-terminal segment (Figure 20-28b). The globular N-terminal segment of type IX collagen extends from the type II fibril at the end of one of its helical segments, as does a chondroitin sulfate GAG chain (chondroitin sulfate is described below) that is sometimes linked to one of the type IX chains. These protruding nonhelical structures are thought to anchor the type II fibril to proteoglycans and other components of the matrix. The interrupted triple-helical structure of type IX and related collagens prevents them from assembling into fibrils, although they can associate with fibrils formed from other collagen types and form covalent cross-links to them.
Mutations affecting type I collagen and its associated proteins cause a variety of human diseases. Certain mutations in the genes encoding the type I collagen α1(I) or α2(I) chains lead to osteogenesis imperfecta, or brittle-bone disease. Because every third position in a collagen α chain must be a glycine for the triple helix to form (see Figure 20-25), mutations of glycine to almost any other amino acid are deleterious, resulting in poorly formed and unstable helices. Only one defective α chain of the three in a collagen molecule can disrupt the whole molecule’s triple-helical structure and function. A mutation in a single copy (allele) of either the α1(I) gene or the α2(I) gene, both located on autosomes, can cause this disorder. Thus it normally shows autosomal dominant inheritance (see Chapter 6).
Absence or malfunctioning of fibril-associated collagen in muscle tissue due to mutations in the type VI collagen genes cause dominant or recessive congenital muscular dystrophies with generalized muscle weakness, respiratory insufficiency, muscle wasting, and muscle-related joint abnormalities. Skin abnormalities have also been reported with type VI collagen disease.