Regulation of Integrin-Mediated Adhesion and Signaling Controls Cell Movement
Cells can exquisitely control the strength of integrin-mediated cell-matrix interactions by regulating integrin’s expression levels, ligand-binding activities, or both. Such regulation is critical to the role of these interactions in cell migration and other functions involving cell movement.
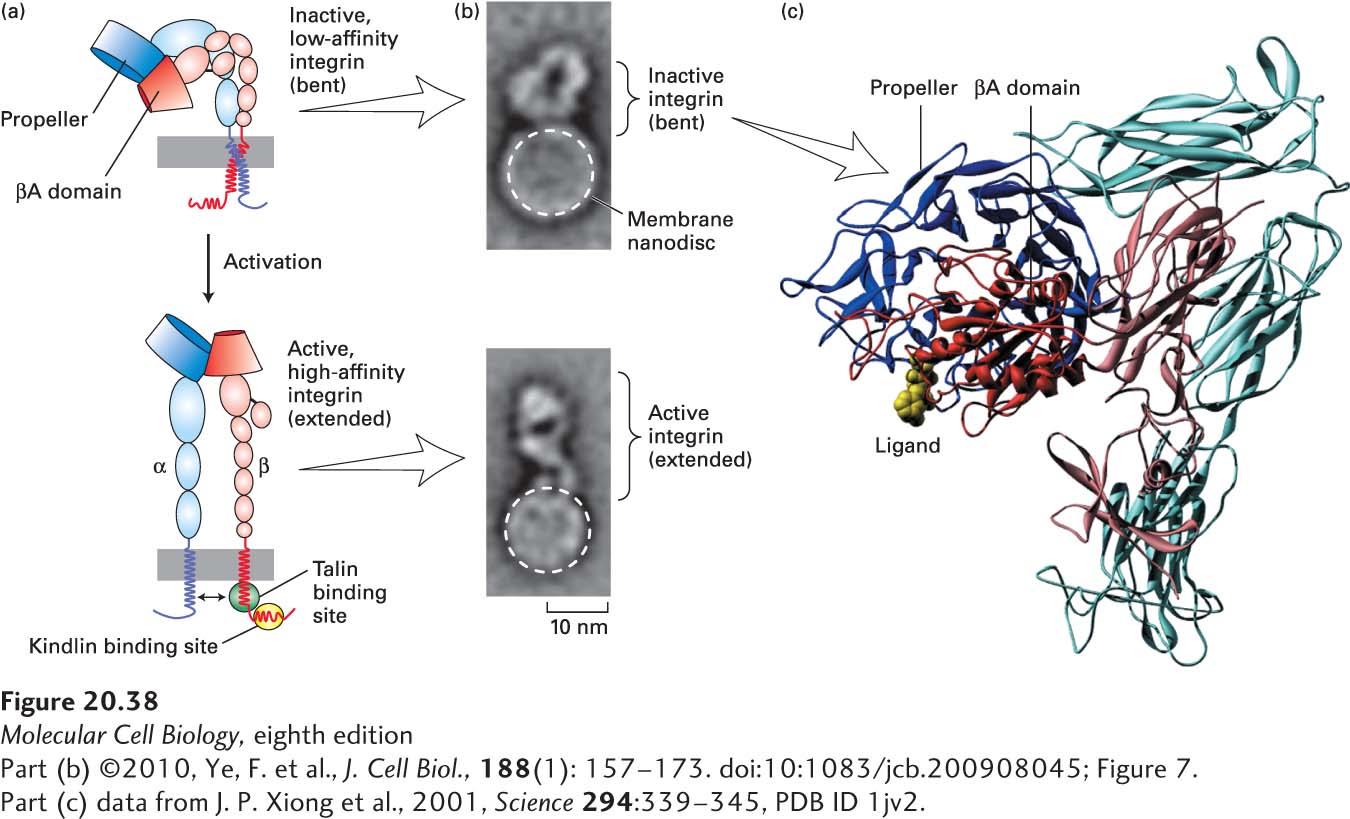
Integrin Binding Many, if not all, integrins can exist in at least two conformations: a low-affinity (inactive) form and a high-affinity (active) form (Figure 20-38a). The results of structural studies and experiments investigating the binding of ligands by integrins have provided a model of the changes that take place when integrins are activated. In the inactive state, the αβ heterodimer is bent (Figure 20-38a, top, and 20-38c), the conformation of the ligand-binding site at the tip of the extracellular domain allows only low-affinity ligand binding, and the transmembrane domains and cytoplasmic C-terminal tails of the two subunits are closely bound together. In the active state, subtle structural alterations in the conformation of the binding site permit tighter (high-affinity) ligand binding and are accompanied by separation of the heterodimer’s transmembrane and cytoplasmic domains (Figure 20-38a, bottom). Activation is also accompanied by the straightening of the molecule into a more extended, linear form in which the ligand-binding site is projected farther away from the surface of the membrane.
EXPERIMENTAL FIGURE 20-38Model for integrin activation. (a) Activation of integrins is thought to be due to conformational changes that include key movements near the propeller and βA domains, which increase the molecule’s affinity for its ligands. These conformational changes are accompanied by straightening of the molecule from the inactive, low-affinity, “bent” conformation (top) to the active, high-affinity, “extended” conformation (bottom). Activation also involves separation (indicated by double-headed arrow, bottom) of the transmembrane and cytoplasmic domains, induced by or resulting in altered interactions with the adapter proteins talin and kindlin, whose sites of binding to the cytoplasmic tail of the β chain are indicated by green and yellow ovals, respectively. (b) Single inactive (bent) integrin αIIbβ3 molecules (top panel) were incorporated into phospholipid nanodiscs (small bilayers in which the extracellular and cytoplasmic domains of the integrin are exposed to a buffer), and the integrin-binding and activating “head” domain of the adapter protein talin was added to some of these preparations (bottom panel). Multiple electron microscopic images of individual nanodiscs were collected and averaged. Phospholipid nanodiscs are indicated by dashed white circles, and the heights of the integrin extracellular regions that extend above the nanodiscs are indicated by brackets. (c) This molecular model of the extracellular region of αvβ3 integrin in its inactive, low-affinity (“bent”) form, with the α subunit in shades of blue and the β subunit in shades of red, is based on x-ray crystallography. The major ligand-binding sites are at the tip of the molecule, where the propeller domain of the α subunit (dark blue) and βA domain (dark red) interact. An RGD peptide ligand is shown in yellow. See M. Arnaout et al., 2002, Curr. Opin. Cell Biol.14:641; R. O. Hynes, 2002, Cell110:673; F. Ye et al., 2010, J. Cell Biol.188:157–173; and M. Moser et al., 2009, Science324:895–899.
These structural models provide an attractive explanation for the ability of integrins to mediate outside-in and inside-out signaling. The binding of certain ECM molecules or CAMs on other cells to the integrin’s extracellular ligand-binding site would hold the integrin in the active form with separated cytoplasmic tails. Intracellular adapter proteins could “sense” the separation of the tails and, as a result, either bind to or dissociate from the tails. The changes in these adapters could then alter the cytoskeleton and activate or inhibit intracellular signaling pathways. Conversely, changes in the metabolic or signaling state of the cells could cause intracellular adapters to bind to or dissociate from the cytoplasmic tails of the integrins and thus force the tails either to separate or to associate (see Figure 20-38a). As a consequence, the integrin would be either bent (inactivated) or straightened (activated), thereby altering its interaction with the ECM or with other cells. Indeed, in vitro studies of purified integrins reconstituted individually into lipid bilayer “nanodiscs” show that binding of the globular “head” domain of the adapter/mechanosensor protein talin (see Figure 20-9b) to the cytoplasmic tail of integrin’s β chain is sufficient to activate integrin, inducing a straightening of the bent conformation into an extended, active form (see Figure 20-38a, bottom; and 20-38b, bottom). Other studies suggest that the efficient activation of integrins in intact cells may also require the participation of another class of adapter proteins called kindlins, which bind to a distinct site on the cytoplasmic tail of integrin’s β chain (see Figure 20-38a, bottom). Kindlin plays a key role in the integrin- and microfibril-mediated activation of TGF-β (inside-out signaling involving elastic fibers and their microfibril-associated protein LTBP, described earlier) and other pathways of integrin-mediated signaling.
Page 964
Platelet function, discussed in more detail below, provides a good example of how cell-matrix interactions are modulated by control of integrin binding activity. Platelets are cell fragments that circulate in the blood and clump together with ECM molecules to form a blood clot. In its basal state, the αIIbβ3 integrin present on the plasma membranes of platelets cannot bind tightly to its protein ligands (including fibrinogen and fibronectin), all of which participate in the formation of a blood clot, because it is in the inactive (bent) conformation. During clot formation, platelets are activated by binding to ECM proteins such as collagen and a large protein called von Willabrand factor that, through binding to receptors, generate intracellular signals. Platelets may also be activated by ADP or the clotting enzyme thrombin. These signals induce changes in signaling pathways within the platelet that result in an activating conformational change in the platelet’s αIIbβ3 integrin. As a consequence, this integrin can bind tightly to extracellular clotting proteins and participate in clot formation. People with genetic defects in the β3 integrin subunit are prone to excessive bleeding, attesting to the role of the αIIbβ3 integrin in the formation of blood clots (see Table 20-4).
Integrin Expression The attachment of cells to ECM components can also be modulated by altering the number of integrin molecules exposed on the cell surface. The α4β1 integrin, which is found on many hematopoietic cells, offers an example of this regulatory mechanism. For these hematopoietic cells to proliferate and differentiate, they must be attached to fibronectin synthesized by supportive (stromal) cells in the bone marrow. The α4β1 integrin on hematopoietic cells binds to a Glu-Ile-Leu-Asp-Val (EILDV) sequence in fibronectin in the ECM, thereby anchoring the cells to the matrix. This integrin also binds to a sequence in a CAM called vascular CAM-1 (VCAM-1), which is present on stromal cells of the bone marrow. Thus hematopoietic cells directly contact the stromal cells as well as the ECM. Late in their differentiation, hematopoietic cells decrease their expression of this integrin; the resulting reduction in the number of α4β1 integrin molecules on the cell surface is thought to allow mature blood cells to detach from the ECM and stromal cells in the bone marrow and enter the circulation.