A Calcium-Binding Protein Regulates Fusion of Synaptic Vesicles with the Plasma Membrane
Fusion of synaptic vesicles with the plasma membrane of axon termini depends on SNAREs, the same type of proteins that mediate membrane fusion of other regulated secretory vesicles, and SM proteins (for Sec1/Munc18-like proteins). The principal v-SNARE in synaptic vesicles (VAMP) tightly binds syntaxin and SNAP-25, the principal t-SNAREs in the plasma membrane of axon termini, to form four-helix SNARE complexes. The assembly of the SNARE complex brings the synaptic vesicle membrane into close proximity to the presynaptic plasma membrane, but the formation of a fusion pore requires an additional step, association of an SM protein with syntaxin. After fusion, proteins within the axon terminus promote disassociation of VAMP from t-SNAREs, as in the fusion of secretory vesicles depicted in Figure 14-10.
Strong evidence for the role of VAMP in neurotransmitter exocytosis is provided by the mechanism of action of botulinum toxin, a bacterial protein that can cause the paralysis and death characteristic of botulism, a type of food poisoning. The toxin is composed of two polypeptides: One binds to motor neurons that release acetylcholine at synapses with muscle cells, facilitating entry of the other polypeptide, a protease, into the cytosol of the axon terminus. The only protein this protease cleaves is VAMP (see Figure 22-26, step 3). After the botulinum protease enters an axon terminus, synaptic vesicles that are not already docked rapidly lose their ability to fuse with the plasma membrane because cleavage of VAMP prevents assembly of SNARE complexes. The resulting block in acetylcholine release at neuromuscular synapses causes paralysis. However, vesicles that are already docked exhibit remarkable resistance to the toxin, indicating that SNARE complexes may already be in a partially assembled, protease-resistant state when vesicles are docked on the presynaptic membrane.
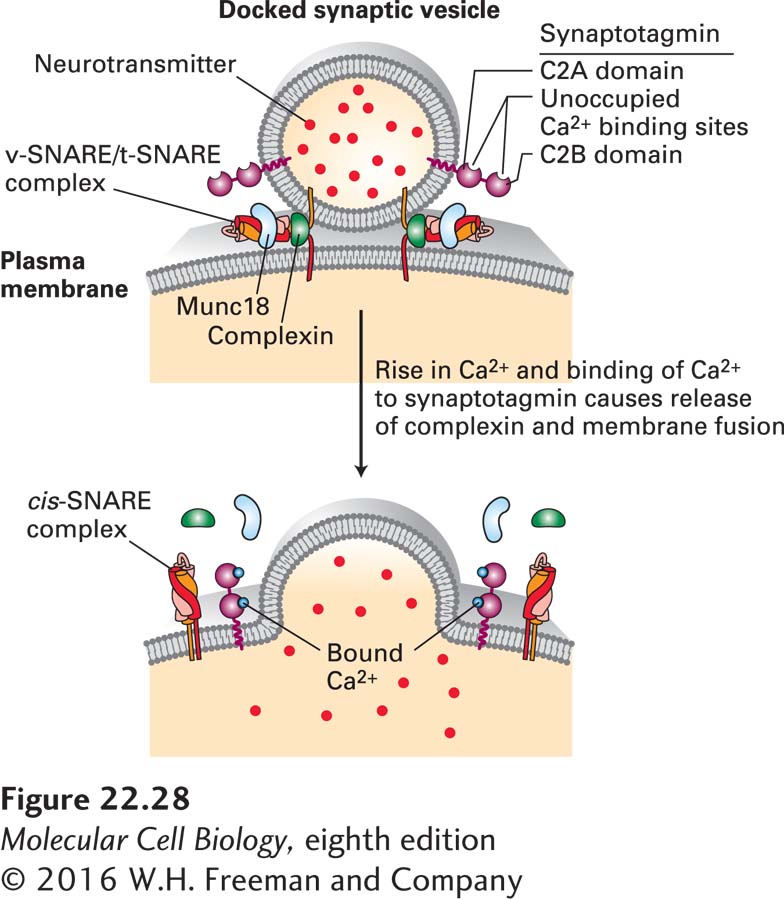
The signal that triggers exocytosis of docked synaptic vesicles is a very localized rise in the Ca2+ concentration in the cytosol near vesicles from 0.1 µM, characteristic of resting cells, to 1–100 µM following arrival of an action potential in stimulated cells. The speed with which synaptic vesicles fuse with the presynaptic membrane after a rise in cytosolic Ca2+ (less than 1 ms) indicates that the fusion machinery is entirely assembled in the resting state and can rapidly undergo a conformational change leading to exocytosis of neurotransmitter (Figure 22-28). A Ca2+-binding protein called synaptotagmin, located in the membrane of synaptic vesicles, is a key component of the vesicle-fusion machinery that triggers exocytosis in response to Ca2+. A protein called complexin is thought to bind to the α-helical bundle of an assembled v-SNARE/t-SNARE complex that bridges the synaptic vesicle and plasma membranes, preventing the final fusion step. Binding of Ca2+ to synaptotagmin relieves this inhibition, releasing complexin and allowing the fusion event to occur very rapidly. While the mechanisms by which synaptotagmin functions are debated, Figure 22-28 depicts a widely accepted model.
Page 1056
FIGURE 22-28Synaptotagmin-mediated fusion of synaptic vesicles with the plasma membrane. Only a few synaptic vesicles are docked at the presynaptic plasma membrane; these are primed for fusion with the plasma membrane. The tight interconnections between the synaptic vesicle and plasma membrane are mediated in part by bundles of four α helices derived from complexes of vesicle v-SNARE and plasma membrane t-SNARE proteins (see Figure 14-10). The fusion of the two membranes is prevented by binding of complexin protein to the v-SNARE/t-SNARE complex. Synaptotagmin is composed of a short intraluminal sequence, a single transmembrane α helix that anchors it in the synaptic vesicle membrane, a linker, and two Ca2+-binding domains termed C2A and C2B. Synaptotagmin without bound Ca2+ may also bind to the v-SNARE/t-SNARE complex and prevent membrane fusion. A localized rise in Ca2+ allows Ca2+ ions to bind to synaptotagmin, altering its three-dimensional conformation. This triggers release of the complexin fusion inhibitor, binding (or altered binding) of synaptotagmin to the v-SNARE/t-SNARE complex, instantaneous membrane fusion, and release of neurotransmitters into the extracellular space. The SM protein Munc18, which binds to syntaxin, is required for SNARE-mediated fusion, although its precise mechanisms of action are not known. See T. Südhof and J. Rothman, 2009, Science323:474 and T. Sudhof, 2013, Neuron80:675–690.
Several lines of evidence support a role for synaptotagmin as the Ca2+ sensor for exocytosis of neurotransmitters. Mutant embryos of Drosophila and C. elegans that completely lack synaptotagmin fail to hatch and exhibit very reduced, uncoordinated muscle contractions. Larvae with partial loss-of-function mutations of synaptotagmin survive, but their neurons are defective in Ca2+-stimulated vesicle exocytosis. Moreover, in mice, mutations in synaptotagmin that decrease its affinity for Ca2+ cause a corresponding increase in the amount of cytosolic Ca2+ needed to trigger rapid exocytosis. Mammals express multiple different synaptotagmin isoforms, each of which has a different binding affinity for Ca2+, and as a result the kinetics of exocytosis depend on the particular synaptotagmin isoform expressed in the neuron.
An important characteristic of synaptic vesicle exocytosis is its speed. Synaptic vesicle fusion occurs within a few hundred microseconds after the arrival of an action potential, which is not very different from the timescale of Ca2+ influx through the voltage-gated Ca2+ channel. What makes this speed possible is the proximity of the release machinery to the voltage-gated Ca2+ channels. This proximity is mediated by two scaffolding proteins called RIM (for Rab3-interacting protein) and RIM-BP (for RIM binding protein), which form a complex between Rab3-containing synaptic vesicles and voltage-gated Ca2+channels. In mice lacking RIM, and flies lacking RIM-BP, active zones lack voltage-gated Ca2+ channels, which leads to a dramatic decrease and desynchronization of neurotransmitter release.