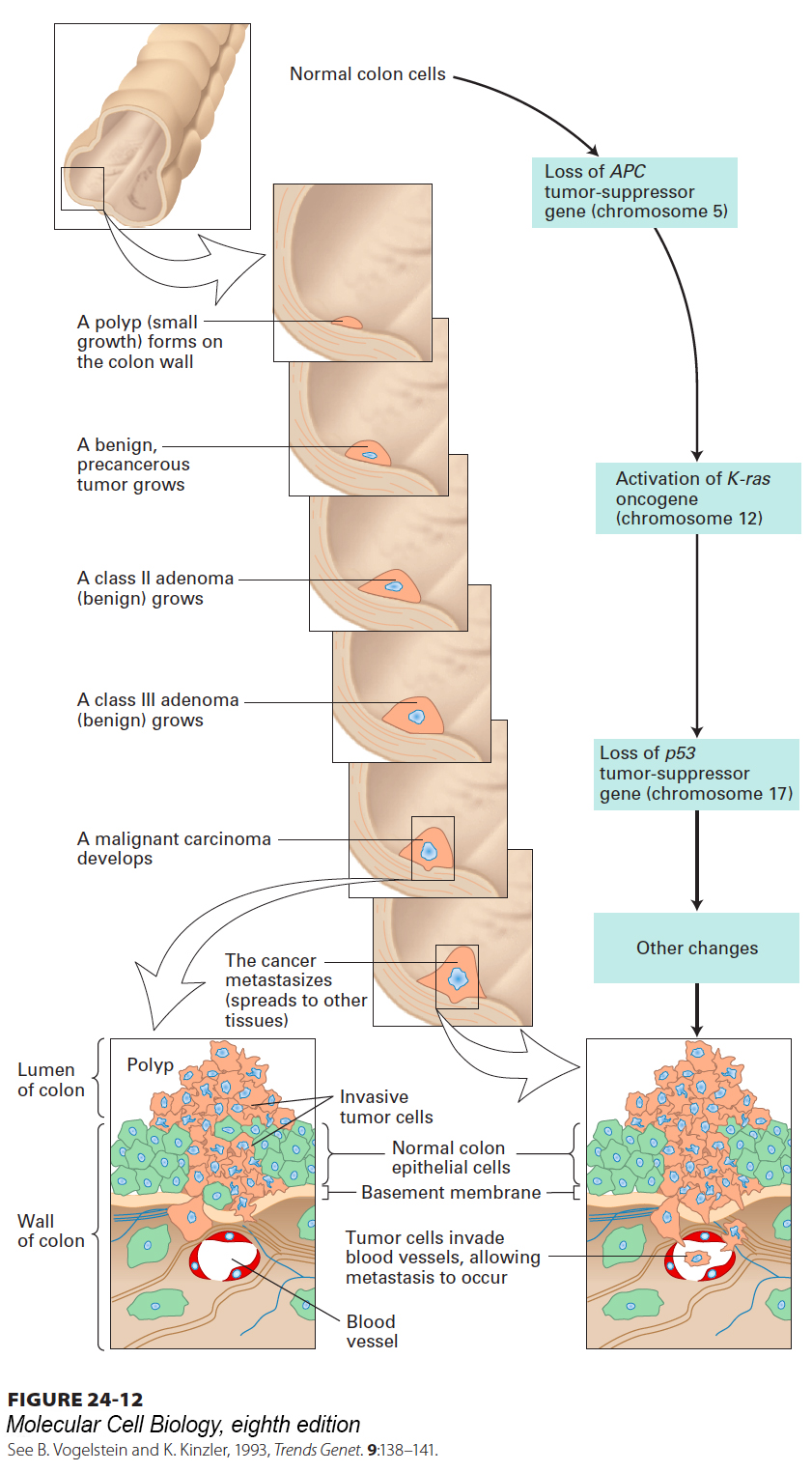
Successive Oncogenic Mutations Can Be Traced in Colon Cancers
Studies on colon cancer provide the most compelling evidence to date for the multi-hit model of cancer induction. Surgeons can obtain fairly pure samples of many human cancers, but since the tumor is observed at only one time, its exact stage of progression cannot be easily determined. An exception is colon cancer, which evolves through distinct, well-characterized morphological stages. Its intermediate stages—polyps, benign adenomas, and carcinomas—can be isolated by a surgeon, allowing mutations that occur in each of these stages to be identified. Numerous studies have shown that colon cancer arises from a series of mutations that commonly occur in a well-defined order, providing strong support for the multi-hit model (Figure 24-12).
FIGURE 24-12The development and metastasis of human colorectal cancer and its genetic basis. A mutation in the APC tumor-suppressor gene in a single epithelial cell causes the cell to divide (although surrounding epithelial cells do not), forming a mass of localized benign tumor cells, called a polyp. Subsequent mutations lead to expression of a constitutively active Ras protein and loss of the tumor-suppressor gene p53. These mutations, together with additional genetic changes yet to be identified, generate a malignant cell. The cell continues to divide, and its progeny invade the basement membrane that surrounds the tissue, but do not penetrate the basement membrane of capillaries (bottom left). Some tumor cells spread into blood vessels that will distribute them to other sites in the body (bottom right). Additional mutations permit the tumor cells to exit from the blood vessels and proliferate at distant sites. See B. Vogelstein and K. Kinzler, 1993, Trends Genet. 9:138–141.
Insight into the progression of colon cancer first came from the study of inherited predispositions to colon cancer such as familial adenomatous polyposis (FAP). Mutations in the Wnt signaling pathway have been identified in many of these syndromes, and it is now believed that deregulation of Wnt signaling results in formation of polyps (precancerous growths) on the inside of the colon wall—not only in people with inherited polyposis syndromes, but also in people afflicted with sporadic (noninherited) forms of colon cancer. The APC (adenomatous polyposis coli) protein is a negative regulator of Wnt signaling (see Chapter 16), which promotes cell cycle entry by activating expression of the MYC gene. The absence of functional APC protein thus leads to inappropriate production of MYC, and cells homozygous for APC mutations proliferate at a rate higher than normal and form polyps. Loss-of-function mutations in the APC gene are the most frequent mutations found in early stages of colon cancer. Most of the cells in a polyp contain the same one or two mutations in the APC gene that result in its loss or inactivation, indicating that they are clones of the cell in which the original mutation occurred. Thus APC is a tumor-suppressor gene, and both alleles of the APC gene must carry an inactivating mutation for polyps to form because cells with one wild-type APC allele express enough APC protein to function normally.
If one of the cells in a polyp undergoes another mutation, this time an activating mutation of the ras gene, its progeny divide in an even more uncontrolled fashion, forming a larger adenoma. Inactivation of the p53 gene follows and results in the gradual loss of normal regulation and the consequent formation of a malignant carcinoma (see Figure 24-12). The p53 protein is a tumor suppressor that halts progression through the cell cycle in response to DNA damage and other stresses. While the three “hits” listed here are certainly crucial parts of the picture, there are likely to be additional contributing genetic events. Not every colon cancer, however, acquires all the later mutations or acquires them in the order depicted in Figure 24-12. Thus different combinations of mutations may result in the same phenotype.
DNA from different human colon carcinomas generally contains mutations in all three genes mentioned here—loss-of-function mutations in the tumor suppressors APC and p53 and an activating (gain-of-function) mutation in the oncogene K-ras (one of the ras family of genes)—establishing that multiple mutations in the same cell are needed for the cancer to form. Some of these mutations appear to confer growth advantages at an early stage of tumor development, whereas other mutations promote the later stages, including invasion and metastasis, which are required for the malignant phenotype. The number of mutations needed for colon cancer progression may at first seem surprising, and might seem to be an effective barrier to tumorigenesis. Our genomes, however, are under constant assault. Recent estimates indicate that sporadically arising polyps have about 11,000 genetic alterations in each cell, though very likely only a few of these alterations are relevant to oncogenesis. The genetic instability that is a hallmark of cancer cells promotes further tumor evolution, allowing for the accelerated creation of tumor cells with increased self-reliance and the ability to metastasize.
Colon carcinoma provides an excellent example of the multi-hit model of cancer. The degree to which this model applies to cancer generally is only now being learned, but it is clear that many types of cancer involve multiple mutations.