Cancer Development Can Be Studied in Cultured Cells and in Animal Models
Most cultured cells have a finite lifespan (see Chapter 4). After about 50 divisions, human cells cease to divide and eventually die due to erosion of their telomeres (see Figure 4-1a). Some cells, however, escape this fate and become immortal; that is, they gain the ability to divide indefinitely. Immortalization is mediated by several kinds of mutations, including loss-of-function mutations in the p19ARF or p53 genes, which are regulators of the cell cycle and cell survival. These mutations allow cells to grow for an unlimited time in culture if they are periodically diluted and supplied with nutrients (see Figure 4-1b).
Page 1147
Page 1148
Immortal cells are not full-blown cancer cells. When they are introduced into an immunocompromised mouse, they fail to form tumors. When further oncogenic mutations are introduced, however, they turn into cancer cells. For example, when a mutant ras gene encoding rasD, a hyperactive form of the Ras protein, is introduced into immortal cells, they are transformed into cancer cells. As we will see in Section 24.3, any gene, such as rasD, that encodes a protein capable of transforming immortalized cells into cancer cells is considered an oncogene. Cell culture experiments have not only provided insights into how oncogenes cause cancer, but have also supported the idea that multiple hits are needed to transform a normal cell into a cancer cell.
Genetically engineered mice have also provided tremendous insights into the steps of tumor initiation and progression. Using mouse models to study cancer is not always straightforward, however. Many tumor-suppressor genes serve essential functions during normal mouse development, so mice lacking both copies of these genes are not viable. The essential functions of these genes during early embryogenesis preclude the study of their role in tumor progression. To circumvent this problem, researchers have begun to employ conditional “knock-in” and “knockout” strategies that allow for the targeted activation or inactivation of a gene in a certain tissue or at a certain stage of development.
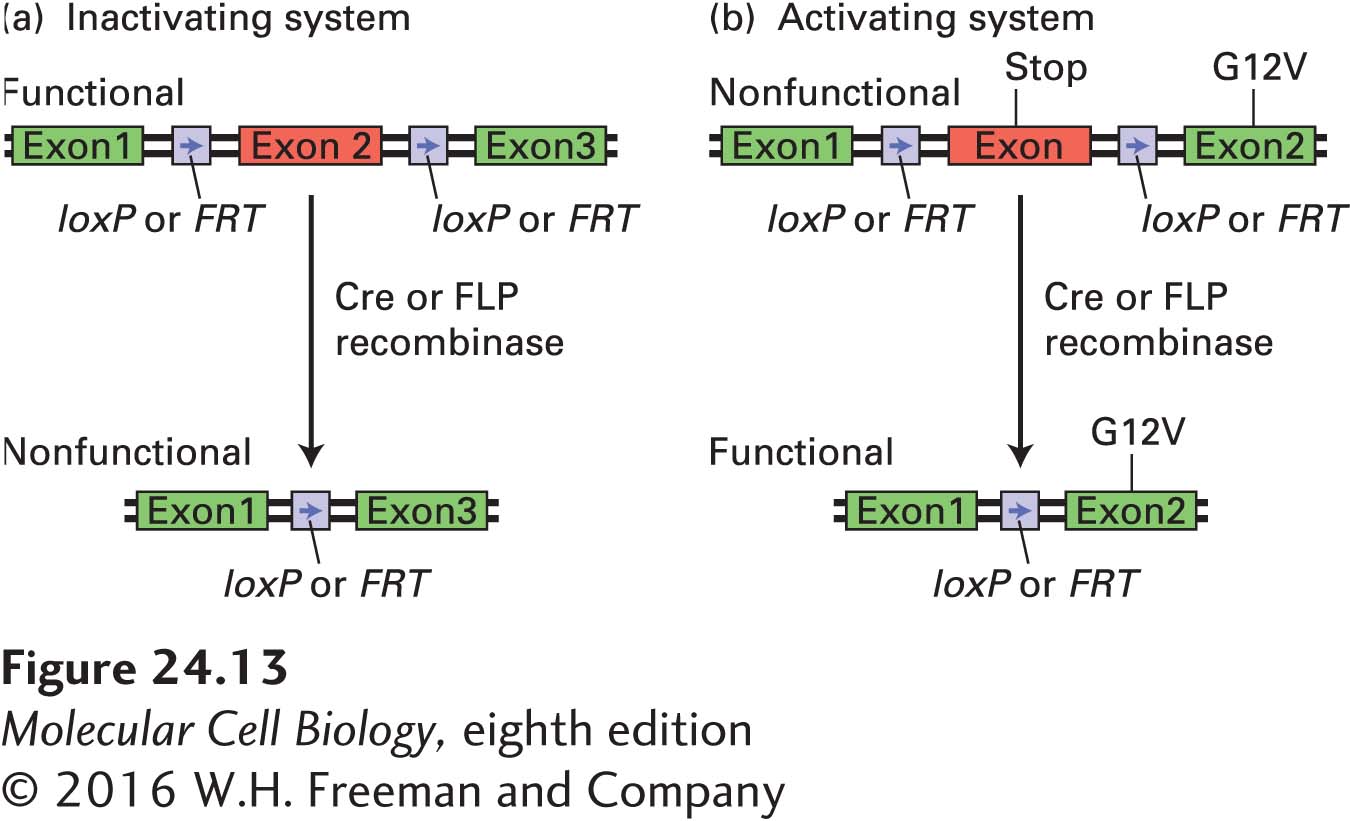
In the conditional mouse model, an allele of a particular oncogene or tumor-suppressor gene is wild type until activated or inactivated with exogenous chemicals or viruses in a tissue- or time-specific manner. At the heart of these conditional systems are the Cre and FLP recombinases. These recombinases facilitate homologous recombination between loxP and FRT sites, respectively (Figure 24-13; see also Figure 6-39). When the recombinases are under the control of a tissue-specific promoter, recombination occurs only in the tissue that produces the recombinase. The recombinase method can be used in two ways. First, the recombinase target sites may flank an exon. Upon induction of the recombinase, that exon is lost and the gene is inactivated (Figure 24-13a). This method is especially useful for inactivating tumor-suppressor genes in a tissue-specific manner. Second, expression of an oncogene can be controlled by introducing into the oncogene an additional exon that contains a stop codon, which makes the gene nonfunctional. However, if the additional exon is flanked by recombinase target sites, the oncogene will be expressed upon induction of the recombinase (Figure 24-13b). Using this system, researchers have examined the role of oncogenic forms of Ras in the mouse and have, using a conditional oncogenic ras allele, created a mouse model of human lung cancer.
FIGURE 24-13Conditional mouse models of cancer. In the inactivating system (a), an exon of interest is flanked by two loxP or FRT sites as shown. Expression of the Cre or FLP recombinase leads to homologous recombination between the two loxP and FRT sites, respectively. This recombination leads to excision of the exon, rendering the gene nonfunctional. In the activating system (b), an additional exon with a stop codon is introduced into the gene of interest, making the gene nonfunctional. This exon is flanked by loxP or FRT sites. When Cre or FLP recombinase is induced, the stop codon–containing exon is recombined out, and the gene of interest is expressed.
The development of promoters that can be regulated by exogenously added chemicals has provided an additional powerful method of controlling gene expression in experimental animals. The most widely used of these methods are the Tet-On and Tet-Off systems. Each system is composed of two parts: the Tet operon promoter, which regulates the expression of the gene of interest, and one of two versions of the transcription factor that binds to the promoter—either the transactivator tTA (in the case of Tet-Off) or the reverse transactivator rtTA (in the case of Tet-On). Both transcription factors bind to the Tet operator to induce gene expression, and both are regulatable by tetracycline or by the tetracycline analog doxycycline, more commonly used by scientists in their experiments. The difference between the two systems lies in the responses of tTA and rtTA to doxycycline binding. Doxycycline inhibits tTA from binding the promoter; thus, in the Tet-Off system, addition of doxycycline turns off transcription. In the Tet-On system, rtTA cannot bind the promoter in the absence of doxycycline, and addition of the drug induces transcription. Doxycycline can be administered by simply adding it to the animals’ water supply. Placing the Tet transcriptional regulators under the control of tissue-specific promoters therefore allows for temporal as well as spatial control of gene expression.
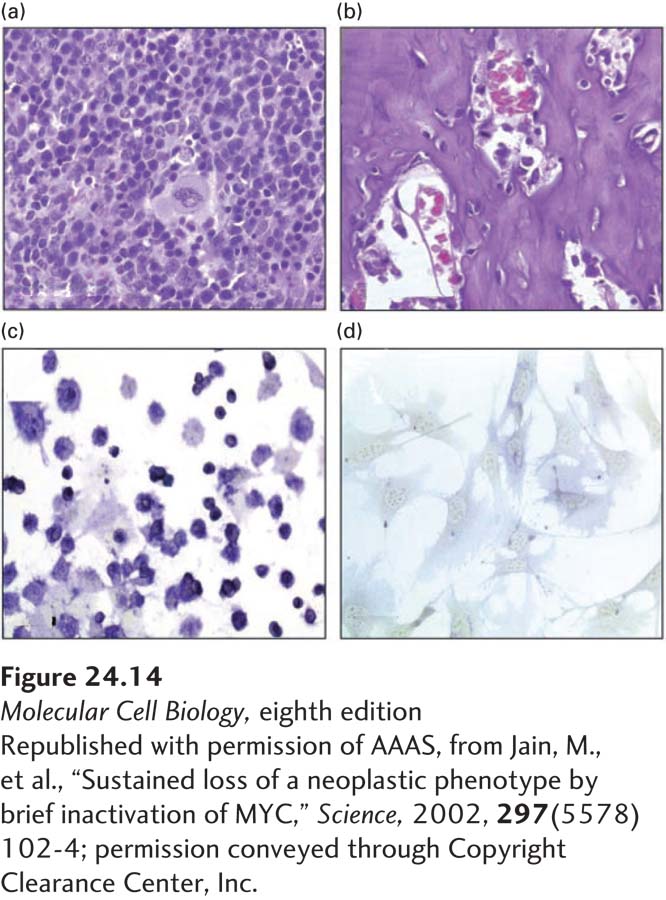
By using the Tet-Off system to control MYC expression, researchers found that survival of a tumor depends on the continuous production of MYC protein. When expression of MYC was even briefly interrupted, osteogenic sarcoma cells ceased dividing and developed into mature osteocytes (Figure 24-14). It is now clear that the continuous activity of oncogenes is required for the survival of many types of tumors. This dependence of tumors on the continuous production of oncogene-encoded proteins, termed oncogene addiction, may provide new opportunities for treatment. Specific inhibitors of these oncogene-encoded proteins—even when applied only transiently—could lead to disease regression.
Page 1149
[Republished with permission of AAAS, from Jain, M., et al., “Sustained loss of a neoplastic phenotype by brief inactivation of MYC,” Science, 2002, 297(5578)102-4; permission conveyed through Copyright Clearance Center, Inc.]
EXPERIMENTAL FIGURE 24-14MYC is continuously needed for tumor growth. Transgenic mice were developed in which MYC expression was driven by the Tet-Off system. One percent of such mice develop osteogenic sarcomas. Wild-type mice were transplanted with osteogenic sarcomas, which causes them to develop the disease. In the transplanted mice, MYC expression was repressed by treating the mice with doxycycline. This treatment caused the osteogenic sarcomas to stop proliferating (a) and differentiate into mature osteocytes (b). After MYC expression was turned off, the tumor cells also lost alkaline phosphatase activity, a marker for osteogenic sarcomas (c, d). Surprisingly, re-expression of MYC protein did not trigger a return to the sarcoma state.
[Republished with permission of AAAS, from Jain, M., et al., “Sustained loss of a neoplastic phenotype by brief inactivation of MYC,” Science, 2002, 297(5578)102-4; permission conveyed through Copyright Clearance Center, Inc.]