Oncogenic Receptors Can Promote Proliferation in the Absence of External Growth Factors
Hyperactivation of a growth-inducing signaling protein due to an alteration of the protein might seem a likely mechanism of cancer, but in fact this rarely occurs. Only one such naturally occurring oncogene, sis, has been discovered. The sis oncogene, which encodes an altered form of platelet-derived growth factor (PDGF), can aberrantly stimulate proliferation of cells that normally express the PDGF receptor when expressed at high levels. A more common event is that cancer cells begin to produce an unaltered growth factor that acts on the cell that produces it. This phenomenon is called autocrine stimulation.
In contrast, oncogenes encoding cell-surface receptors that transduce growth-promoting signals have been associated with several types of cancer. Many of these receptors have intrinsic protein tyrosine kinase activity in their cytosolic domains, which is quiescent until activated. Ligand binding to the external domains of these receptor tyrosine kinases (RTKs) leads to their dimerization and activation of their kinase activity, initiating an intracellular signaling pathway that ultimately promotes proliferation.
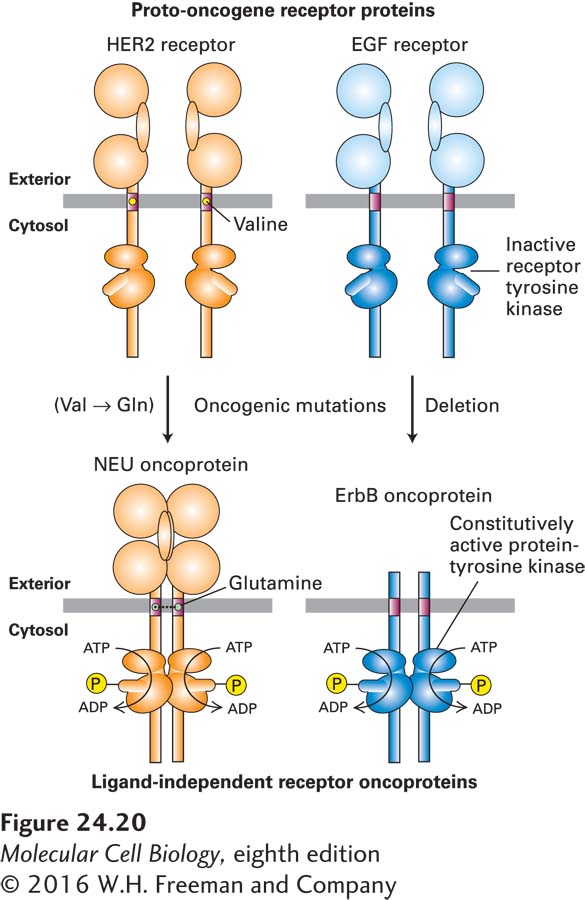
In some cases, a point mutation changes a normal RTK into one that dimerizes and is constitutively active even in the absence of ligand. For instance, a single point mutation converts the normal human EGF receptor 2 (HER2) into the NEU oncoprotein (“NEU” for its first known role, in neuroblastoma), which is an initiator of certain mouse cancers (Figure 24-20, left). Similarly, human tumors called multiple endocrine neoplasia type 2 produce a constitutively active dimeric glia-derived neurotrophic factor (GDNF) receptor that results from a point mutation in the extracellular domain. The GDNF receptor and the HER2 receptor are both protein tyrosine kinases, so the constitutively active forms excessively phosphorylate their downstream target proteins. In other cases, deletion of much of the extracellular ligand-binding domain produces a constitutively active oncogenic receptor. For example, deletion of the extracellular domain of the normal EGF receptor (Figure 24-20, right) converts it to the dimeric ErbB oncoprotein (from erythroblastosis virus, in which a viral version of the altered gene was first identified). Mutations leading to overproduction of a normal RTK can also be oncogenic. For instance, many human breast cancers overproduce a normal HER2 receptor because of amplification of its encoding gene. As a result, the cells are stimulated to proliferate in the presence of very low concentrations of EGF and related hormones, concentrations too low to stimulate proliferation of normal cells (see Chapter 16).
Page 1160
FIGURE 24-20Effects of oncogenic mutations in proto-oncogenes that encode cell-surface receptors.Left: A mutation that alters a single amino acid (valine to glutamine) in the transmembrane region of the HER2 receptor causes dimerization of the receptor, even in the absence of the normal EGF-related ligand, transforming it into the oncoprotein NEU, a constitutively active kinase. Right: A deletion that causes loss of the extracellular ligand-binding domain in the EGF receptor leads, for unknown reasons, to constitutive activation of the kinase activity of the resulting oncoprotein, ErbB.