The protein p53 is a central player in tumorigenesis. It is thought that most, if not all, human tumors have mutations either in p53 itself or in proteins that regulate p53 activity. Cells with functional p53 become arrested in G1 when exposed to DNA-damaging irradiation, whereas cells lacking functional p53 do not. Unlike other cell cycle proteins, p53 is present at very low levels in normal cells because it is extremely unstable and rapidly degraded. Mice lacking p53 are largely viable and healthy except for a predisposition to develop multiple types of tumors.
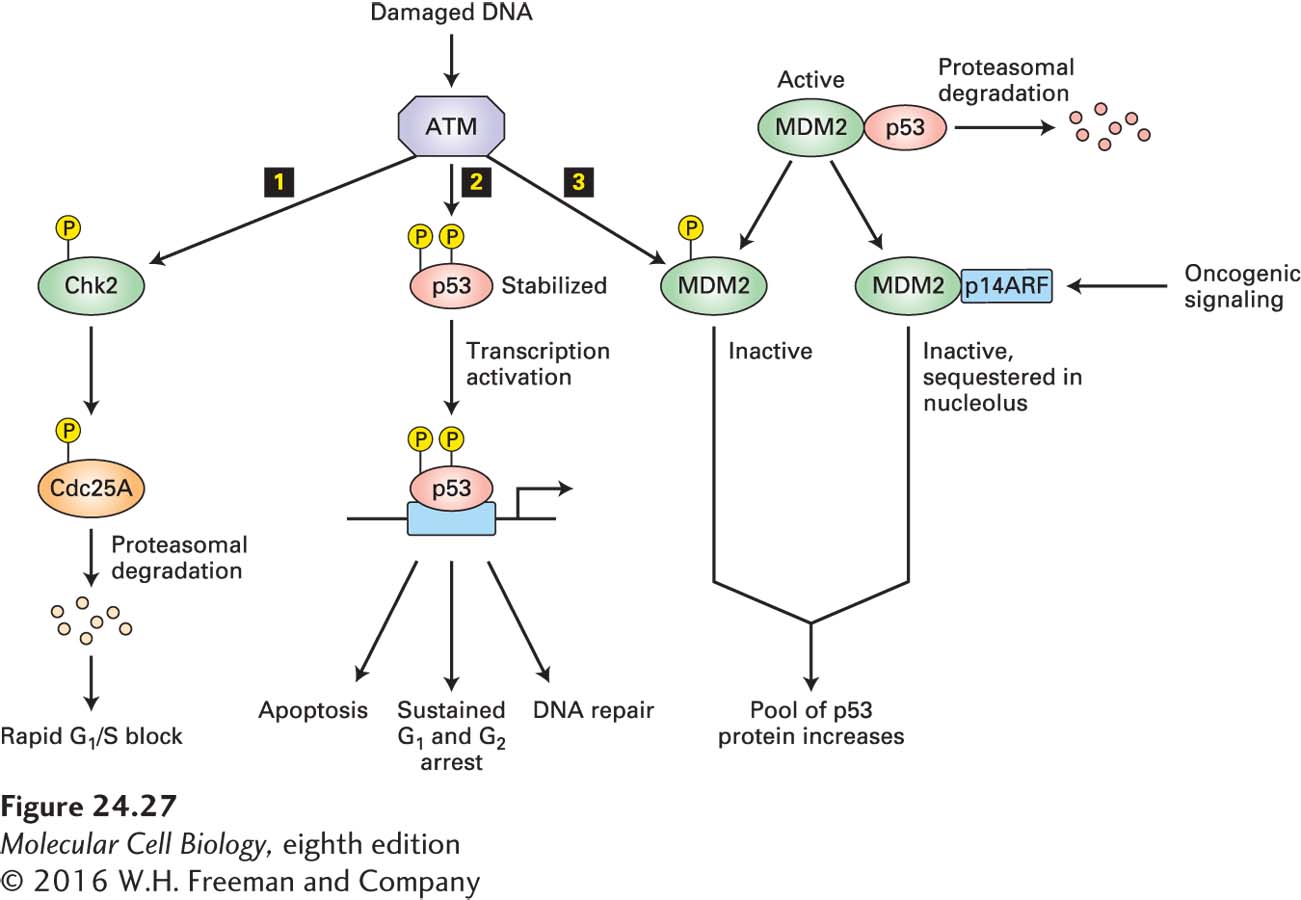
In normal mice, the amount of p53 protein is heightened—a post-transcriptional response—only in stressful situations such as exposure to UV or γ-irradiation, heat, or hypoxia. Irradiation by γ-rays creates lesions in the DNA. Serine kinases ATM or ATR are recruited to these sites of damage and are activated. They then phosphorylate p53 on a serine residue in the N-terminus of the protein. This phosphorylation causes the protein to evade ubiquitin-mediated degradation, leading to a marked increase in its concentration (Figure 24-27). The stabilized p53 activates transcription of the gene encoding p21, which binds to and inhibits mammalian cyclin E-CDK2. As a result, cells with damaged DNA are arrested in G1, allowing time for DNA repair by the mechanisms discussed in Chapter 5, or permanently arrested—that is, they become senescent. The activity of p53 is not limited to inducing cell cycle arrest, however. In addition, this multipurpose tumor suppressor stimulates production of pro-apoptotic proteins (as we will see shortly) and DNA-repair enzymes (see Figure 24-27). Senescence and apoptosis may in fact be the most important means through which p53 prevents tumor growth.
FIGURE 24-27Arrest in G1in response to DNA damage. The kinase activity of ATM is activated in response to DNA damage due to various stresses (e.g., UV irradiation, heat). Activated ATM then triggers three pathways leading to arrest in G1: 1 Chk2 is phosphorylated and, in turn, phosphorylates Cdc25A, thereby marking it for degradation and blocking its role in CDK2 activation. 2 In a second pathway, phosphorylation of p53 stabilizes it, permitting p53-activated expression of genes encoding proteins that cause arrest in G1, promote apoptosis, or participate in DNA repair. 3 The third pathway is another way of controlling the pool of p53. The MDM2 protein in its active form can form a complex with p53, inhibiting the transcription factor and causing p53 ubiquitinylation and subsequent proteasomal degradation. ATM phosphorylates MDM2 to inactivate it, causing increased stabilization of p53. In addition, MDM2 levels are controlled by p14ARF (p19ARF in the mouse), which binds MDM2 and sequesters it in the nucleolus, where it cannot access p53. The p14ARF gene is induced by high levels of mitogenic signaling, which are frequently observed in cells carrying oncogenic mutations in growth factor signaling pathways. The human MDM2 gene is frequently amplified in sarcomas, which presumably causes excessive inactivation of p53. Similarly, p14ARF is also found mutated in some cancers.
Page 1166
The activity of p53 is normally kept low by a protein called MDM2. When MDM2 is bound to p53, it inhibits the transcription-activating ability of p53 and at the same time, because it has E3 ubiquitin ligase activity, catalyzes the ubiquitinylation of p53, thus targeting it for proteasomal degradation. Phosphorylation of p53 by ATM or ATR displaces bound MDM2 from p53, thereby stabilizing it. Because the MDM2 gene is itself transcriptionally activated by p53, MDM2 functions in an autoregulatory feedback loop with p53, perhaps normally preventing excess p53 function. The MDM2 gene is amplified in many sarcomas and other human tumors that contain a normal p53 gene. Even though functional p53 is produced by such tumor cells, the elevated MDM2 levels reduce the p53 concentration enough to abolish the p53-induced arrest in G1 in response to irradiation. A key regulator of MDM2 is the p14ARF protein, encoded by the multi-tumor-suppressor locus that also encodes the INK4 proteins. The p14ARF protein binds to MDM2 and sequesters it in the nucleolus, away from p53. Normal p14ARF levels are so low in tissues that the protein is barely detectable—otherwise, it would cause p53 accumulation and hence cell cycle arrest or apoptosis. However, in response to oncogenic signaling—that is, in the presence of high levels of pro-proliferation signals—p14ARF transcription is induced by the E2F transcription factor. Thus p14ARF is an important inhibitor of tumorigenesis, since it induces p53 activation when pro-proliferation signaling reaches unphysiologically high levels through hyperactivating mutations in the signaling pathways. For pro-proliferation signaling pathways to cause uncontrolled proliferation, as is seen in cancer, this p53 up-regulation must not occur. It is therefore not surprising that p53 is inactive in most human tumors through loss of p53 function itself, down-regulation of positive regulators of p53 function such as p14ARF, or up-regulation of negative regulators of p53 such as MDM2.
The activity of p53 is also inhibited by a human papillomavirus (HPV) protein called E6. HPV encodes two proteins that contribute to its ability to induce stable transformation and mitosis in a variety of cultured cells. These proteins, E6 and E7, bind to and inhibit the p53 and RB tumor suppressors, respectively. Acting together, E6 and E7 are sufficient to induce transformation in the absence of mutations in cell proliferation regulatory proteins.
The active form of p53 is a tetramer of four identical subunits. A missense point mutation in one of the two p53 alleles in a cell can abrogate almost all p53 activity because virtually all the oligomers will contain at least one defective subunit, and such oligomers have reduced ability to activate transcription. Oncogenic p53 mutations thus act in a dominant-negative manner, in which a single mutant allele causes a loss of function. The loss of function is incomplete, so in order to grow more rapidly, tumor cells still sometimes lose the remaining functional allele (loss of heterozygosity). As we learned in Chapter 6, dominant-negative mutations can occur in proteins whose active forms are multimeric or whose function depends on interactions with other proteins. In contrast, loss-of-function mutations in other tumor-suppressor genes (e.g., RB) are recessive because the encoded proteins function as monomers and mutation of a single allele has little functional consequence.
The p53 protein is a key defense mechanism against oncogenic transformation. This is best illustrated by the observation that loss-of-function mutations in the p53 gene occur in more than 50 percent of human cancers. What does p53 protect us against? Unlike Rb, which prevents inappropriate proliferation, p53 guards the cell from genetic changes. When the p53 G1 checkpoint control does not operate properly, damaged DNA can replicate, generating mutations and DNA rearrangements that are passed on to daughter cells and make their transformation into metastatic cells more likely. For example, loss of p53 function leads to a hundredfold or greater increase in the frequency of gene amplification. At the same time, loss of p53 function prevents apoptosis, contributing to evolution of transformed cells. Because of its central role in preventing tumorigenesis, researchers are intensely searching for compounds that can restore p53 function as a new way of treating a broad spectrum of human tumors.