Loss of DNA-Repair Systems Can Lead to Cancer
As our previous discussion has shown, alterations in DNA that lead to the malfunction of tumor-suppressor proteins and the production of oncoproteins are the underlying cause of most cancers. These oncogenic mutations in key growth and cell cycle regulatory genes include insertions, deletions, and base substitutions as well as chromosomal amplifications and translocations. Damage to DNA-repair systems (see Chapter 5) leads to an increased rate of these genetic alterations. Of the many mutations that accumulate in cells with defects in DNA repair mechanisms, some affect cell cycle regulators, some cell adhesion, and some the ability to migrate through basement membranes, discussed at the beginning of this chapter. Cells that have accumulated these kinds of mutations may become cancerous. Furthermore, some DNA-repair mechanisms themselves are error prone (see Figure 6-37). Those errors also contribute to oncogenesis. The inability of tumor cells to maintain genomic integrity leads to the formation of a heterogeneous population of malignant cells. For this reason, chemotherapy directed toward a single gene, or even a group of genes, is likely to be ineffective in wiping out all malignant cells. This problem adds to the interest in therapies that interfere with the blood supply to tumors, target aneuploid cells, or in other ways act on multiple types of tumor cells.
Page 1167
Normal dividing cells usually employ several mechanisms to prevent the accumulation of detrimental mutations that could lead to cancer. One form of protection against mutation for stem cells is their relatively low rate of division, which reduces the possibility of DNA damage incurred during DNA replication and mitosis. Furthermore, the progeny of stem cells do not have the ability to divide indefinitely. After several rounds of division, they exit the cell cycle, reducing the possibility of mutation-induced misregulation of cell division associated with dangerous tumors. Furthermore, if multiple mutations are required for a tumor to grow, attract a blood supply, invade neighboring tissues, and metastasize, a low rate of replication combined with the normal low rate of mutations (10−9) provides further shielding from cancer. However, these safeguards can be overcome if a powerful mutagen reaches the cells, or if DNA repair is compromised and the mutation rate rises. When cells with stem cell-like growth properties are mutated by environmental poisons and are unable to efficiently repair the damage, cancer can occur.
Even without exposure to any external carcinogens or mutagens, normal biological processes generate a large amount of DNA damage. That damage is due to depurination reactions, to alkylation reactions, and to the generation of reactive species such as oxygen radicals, all of which alter DNA. It has been estimated that in every cell, more than 20,000 alterations to the DNA occur each day from reactive oxygen species and depurination. Thus DNA repair is a crucial defense system against genetic change, and hence against cancer.
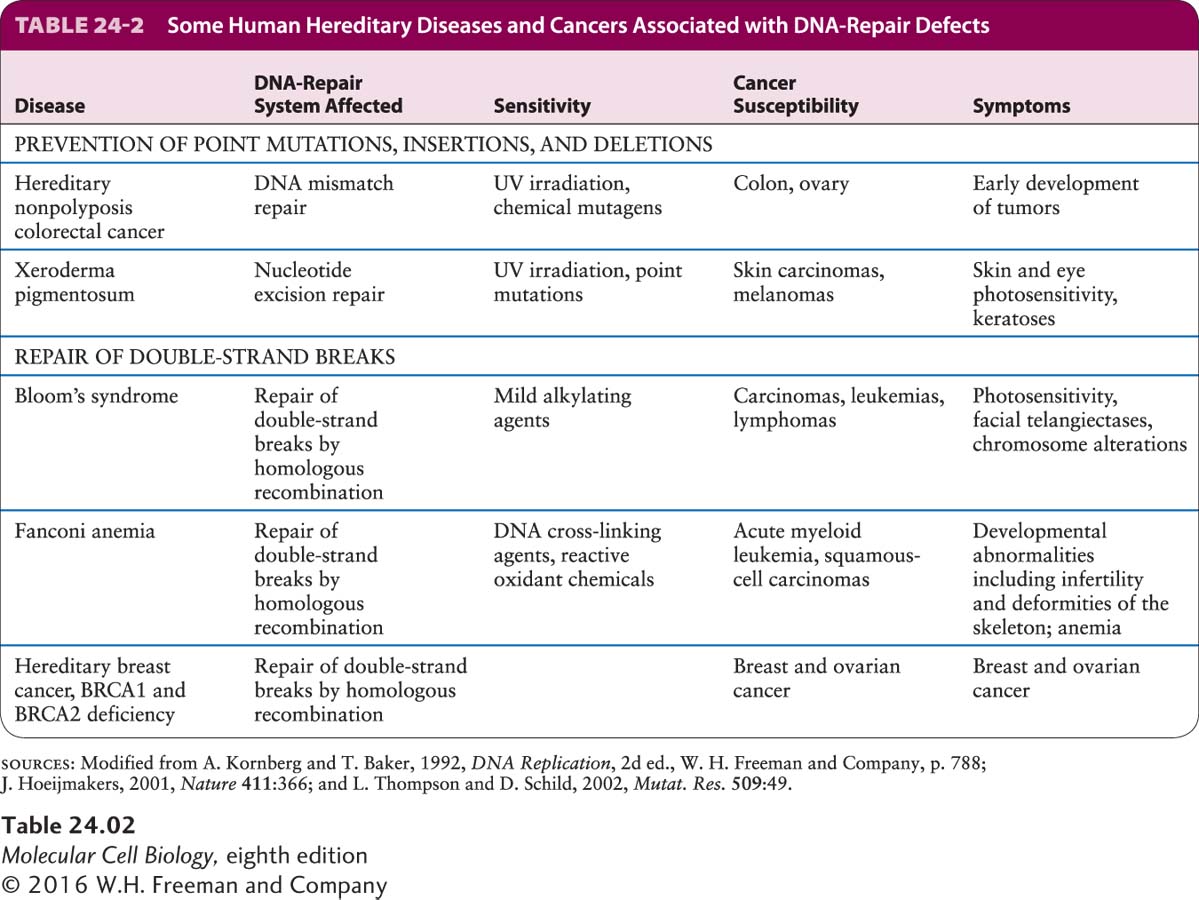
The normal role of genome maintenance genes is to prevent or repair DNA damage. Loss of the high-fidelity DNA-repair systems that are described in Chapter 5 correlates with increased risk for cancer. For example, people who inherit mutations in genes that encode a crucial mismatch-repair or excision-repair protein have an enormously increased probability of developing certain cancers (Table 24-2). Without proper DNA repair, people with xeroderma pigmentosum (XP) or hereditary nonpolyposis colorectal cancer (HNPCC, also known as Lynch syndrome) have a propensity to accumulate mutations in many other genes, including those that are critical in controlling cell growth and proliferation. XP causes affected people to develop skin cancer at about a thousand times the normal rate. Seven of the eight known XP genes encode components of the excision-repair machinery, and in the absence of this repair mechanism, genes that control the cell cycle or otherwise regulate cell growth and death become mutated. HNPCC genes encode components of the mismatch-repair system, and mutations in these genes are found in 20 percent of sporadic colon cancers. The cancers progress from benign polyps to full-fledged tumors much more rapidly than usual, presumably because the initial cancer cells are undergoing continuous mismatch mutagenesis without repair.
Page 1168
One gene frequently mutated in colon cancers because of the absence of mismatch repair encodes the type II receptor for TGF-β (see Figure 24-24). The gene encoding this receptor contains a sequence of 10 adenines in a row. Because of “slippage” of DNA polymerase during replication, this sequence often undergoes mutation to a sequence containing 9 or 11 adenines. If the mutation is not fixed by the mismatch-repair system, the resultant frameshift in the protein-coding sequence abolishes production of the normal receptor protein. As noted earlier, such inactivating mutations make cells resistant to growth inhibition by TGF-β, thereby contributing to the unregulated growth characteristic of these tumors. This finding attests to the importance of mismatch repair in correcting genetic damage that might otherwise lead to uncontrolled cell proliferation.
All DNA-repair mechanisms use a family of DNA polymerases different from the standard Pol α, Pol δ, and Pol ε replicative DNA polymerases to correct DNA damage. Nine of these polymerases, including one called DNA polymerase β, are capable of using templates that contain DNA adducts and other chemical modifications, even missing bases. These enzymes are called lesion-bypass DNA polymerases. Each member of this polymerase family has distinct capabilities to cope with particular types of DNA lesions. Presumably such polymerases are tolerated because often any repair is better than none. They are the polymerases of last resort, the ones used when more conventional and accurate polymerases are unable to perform, and they carry out a mutagenic replication process. DNA Pol β does not proofread and is overexpressed in certain tumors, perhaps because it is needed at high levels for cells to be able to divide at all in the face of a growing burden of mutations. Error-prone repair systems are thought to mediate much, if not all, of the carcinogenic effect of chemicals and radiation, since it is only after the repair that a heritable mutation exists. There is growing evidence that mutations in DNA Pol β are associated with tumors. When 189 tumors were examined, 58 had mutations in the DNA Pol β gene, and most of these mutations were found neither in normal tissue from the same patient nor in the normal spectrum of mutations found in different people. Expressing two of the mutant polymerase forms in mouse cells caused them to grow with a transformed appearance and an ability to form foci.
Double-strand breaks are particularly severe lesions because incorrect rejoining of double strands of DNA can lead to gross chromosomal rearrangements and translocations, such as those that produce a hybrid gene or bring a growth regulatory gene under the control of a different promoter or enhancer. Often the repair of such damage depends on using the homologous chromosome as a guide (see Figure 6-39). The B and T cells of the immune system are particularly susceptible to DNA rearrangements caused by double-strand breaks created during rearrangement of their immunoglobulin or T-cell receptor genes, which explains the frequent involvement of these loci in leukemias and lymphomas. BRCA1 and BRCA2, genes implicated in human breast and ovarian cancers, encode important components of DNA-break repair systems. Cells lacking either of the BRCA functions are unable to repair DNA where the homologous chromosome is providing the template for repair.