Advanced Techniques in Mass Spectrometry Are Critical to Proteomic Analysis
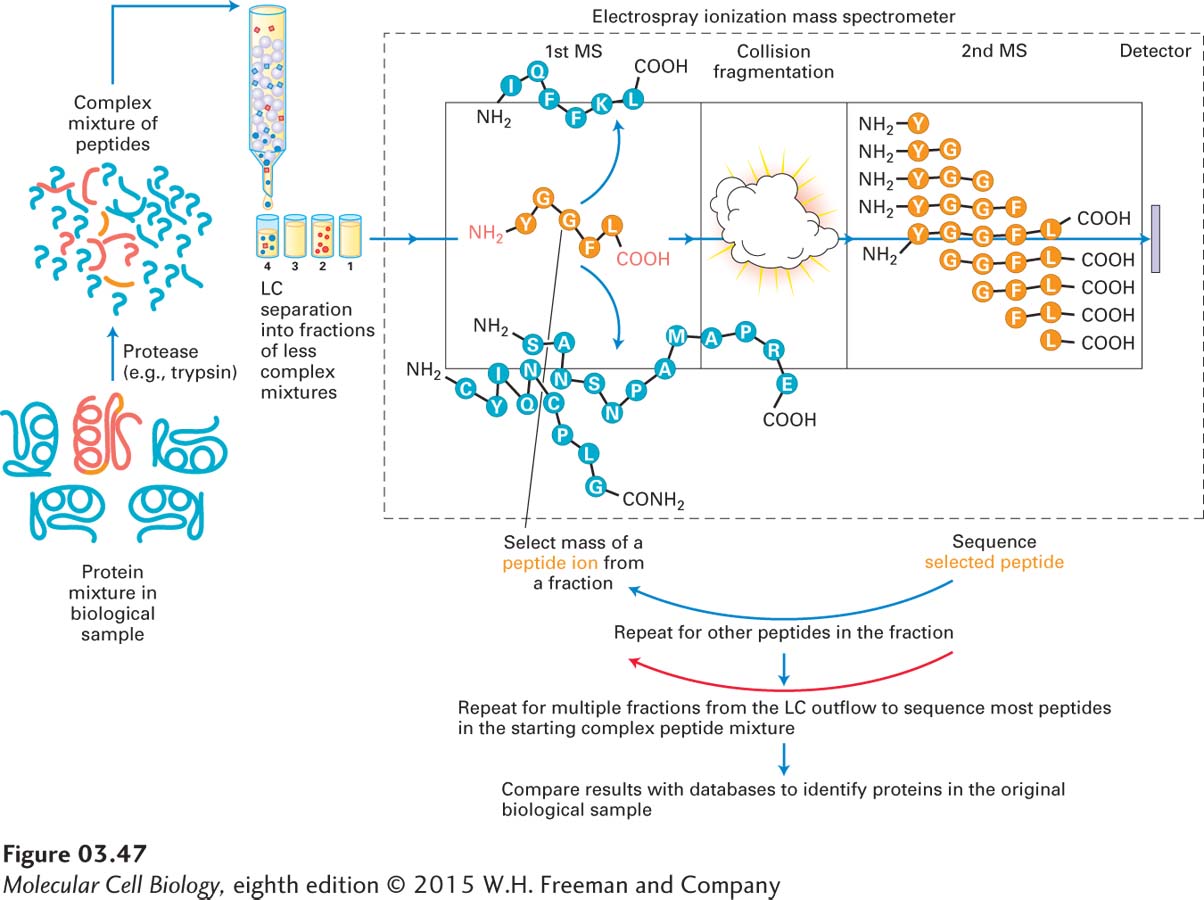
Advances in proteomics technologies (e.g., mass spectrometry) profoundly affect the types of questions that can be practically studied. For many years, two-dimensional gel electrophoresis allowed researchers to separate, display, and characterize complex mixtures of proteins (see Figure 3-39). The spots on a two-dimensional electrophoresis gel could be excised, the protein fragmented by proteolysis (e.g., by trypsin digestion), and the fragments identified by MS. An alternative to this two-dimensional gel electrophoresis method is high-throughput LC-MS/MS. Figure 3-47 outlines the general LC-MS/MS approach, in which a complex mixture of proteins is digested with a protease; the myriad resulting peptides are fractionated by LC into multiple, less complex fractions, which are slowly but continuously injected by electrospray ionization into a tandem mass spectrometer. The fractions are then sequentially subjected to multiple cycles of MS/MS until the sequences of many of the peptides have been determined and used to identify the proteins in the original biological sample. Detection of a substantial fraction of the proteins in whole cells or tissues currently requires samples containing more than 50 µg of protein, an amount equivalent to the protein content of some 70,000–200,000 mammalian cells. Efforts are under way to increase the sensitivity of the method so that eventually one might be able to analyze the proteome of an individual cell.
EXPERIMENTAL FIGURE 3-47LC-MS/MS is used to identify the proteins in a complex biological sample. A complex mixture of proteins in a biological sample (e.g., an isolated preparation of Golgi organelles) is digested with a protease. The mixture of resulting peptides is fractionated by liquid chromatography (LC) into multiple, less complex, fractions, which are slowly but continuously injected by electrospray ionization into a tandem mass spectrometer. The fractions are then sequentially subjected to multiple cycles of MS/MS until the masses and sequences of many of the peptides have been determined and used to identify the proteins in the original biological sample through comparison with protein databases.
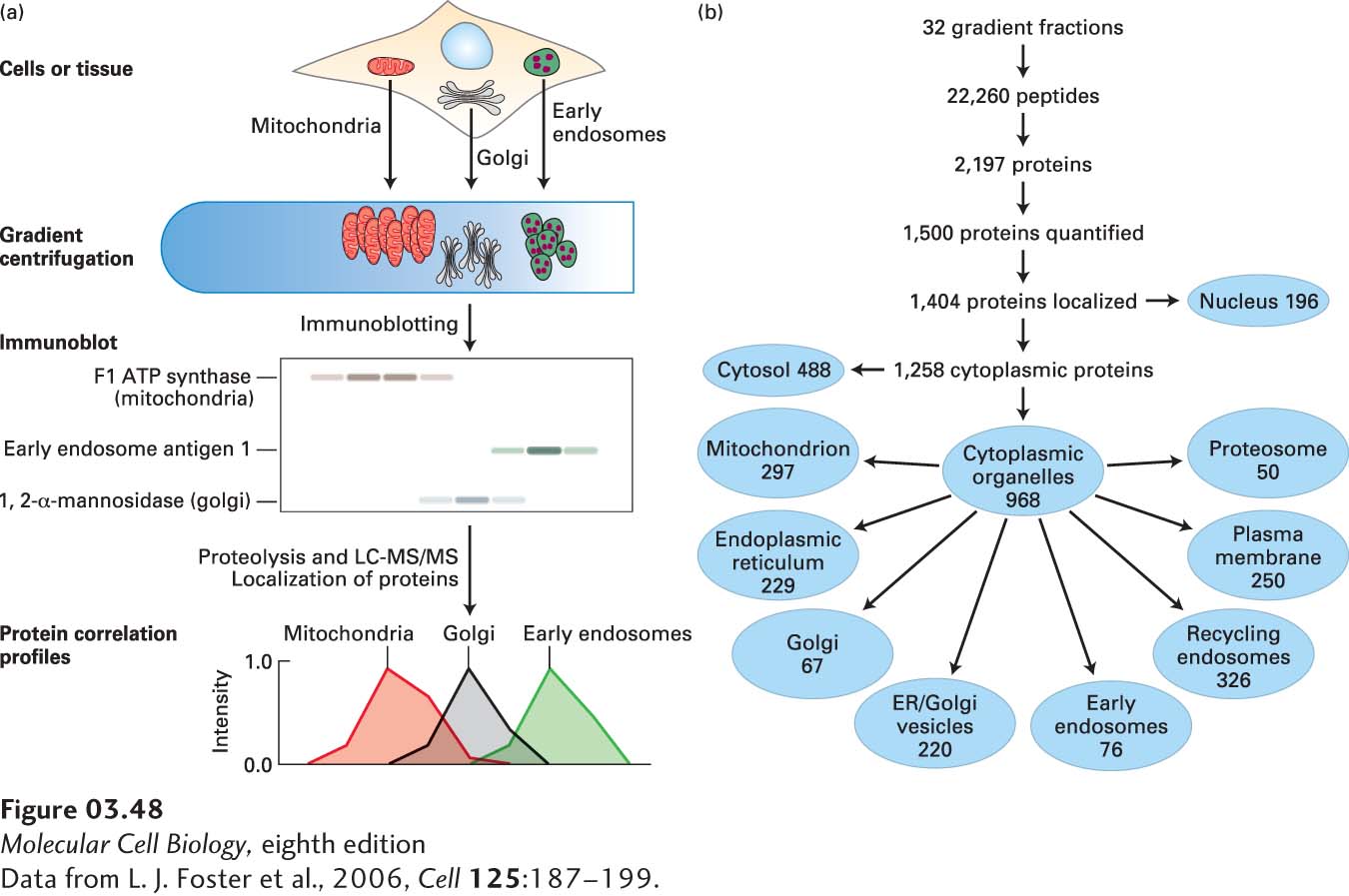
An example of the use of LC-MS/MS to identify many of the proteins in each organelle is seen in Figure 3-48. Cells from murine (mouse) liver tissue were mechanically broken to release the organelles, and the organelles were partially separated by density-gradient centrifugation. The locations of the organelles in the gradient were determined using immunoblotting with antibodies that recognized previously identified, organelle-specific proteins. Fractions from the gradient were then subjected to LC-MS/MS to identify the proteins in each fraction, and the distributions in the gradient of many individual proteins were compared with the distributions of the organelles. This strategy permitted the assignment of many individual proteins to one or more organelles (organelle proteome profiling). More recently, a combination of organelle purification, MS, biochemical localization, and computational methods has been used to show that at least a thousand distinct proteins are localized in the mitochondria of humans and mice.
[Data from L. J. Foster et al., 2006, Cell125:187–199.]
EXPERIMENTAL FIGURE 3-48Density-gradient centrifugation and LC-MS/MS can be used to identify many of the proteins in organelles. (a) The cells in liver tissue were mechanically broken to release the organelles, and the organelles were partially separated by density-gradient centrifugation. The locations of the organelles—which were spread out through the gradient and somewhat overlapped with one another—were determined using immunoblotting with antibodies that recognized previously identified, organelle-specific proteins. Fractions from the gradient were subjected to proteolysis and LC-MS/MS to identify the peptides, and hence the proteins, in each fraction. Comparisons with the locations of the organelles in the gradient (called protein correlation profiling) permitted assignment of many individual proteins to one or more organelles (organelle proteome identification). (b) The hierarchical breakdown of data derived from the procedures in part (a). Note that not all proteins identified could be assigned to organelles and that some proteins were assigned to more than one organelle.
[Data from L. J. Foster et al., 2006, Cell125:187–199.]
Page 124
Proteomics methods combined with molecular genetics methods are currently being used to identify all the protein complexes in eukaryotic cells. For example, in the yeast Saccharomyces cerevisiae, approximately 500 complexes, with an average of 4.9 distinct proteins per complex, have been identified. These complexes, in turn, are involved in at least 400 complex-to-complex interactions. Such systematic proteomic studies are providing new insights into the organization of proteins within cells and into how proteins work together to permit cells to live and function.
Phosphoproteomics, the identification and quantification of phosphorylation sites on the proteins in a complex mixture, is playing a growing role in the analysis of cell metabolism and regulation. As we have already learned, the reversible phosphorylation of proteins by kinases and phosphatases is a key mechanism for regulating proteins in cells. Phosphoproteomics permits the simultaneous determination of the phosphorylation states of many proteins and thus provides an important tool for analyzing complex cellular regulatory networks. Only a fraction—in some cases, only a small fraction—of a particular protein might be phosphorylated. Thus phosphoproteomic analysis can require 50–100 times more initial cell or tissue sample material (from about 2.5 to more than 20 mg of total cellular protein per sample) than does standard proteomic analysis. As a consequence, investigators usually use affinity chromatography methods with either metal-containing (e.g., Fe3+ or TiO2) or antibody-containing columns to separate phosphopeptides from nonphosphorylated peptides (phosphopeptide enrichment) prior to subjecting the phosphopeptides to LC-MS/MS analysis.