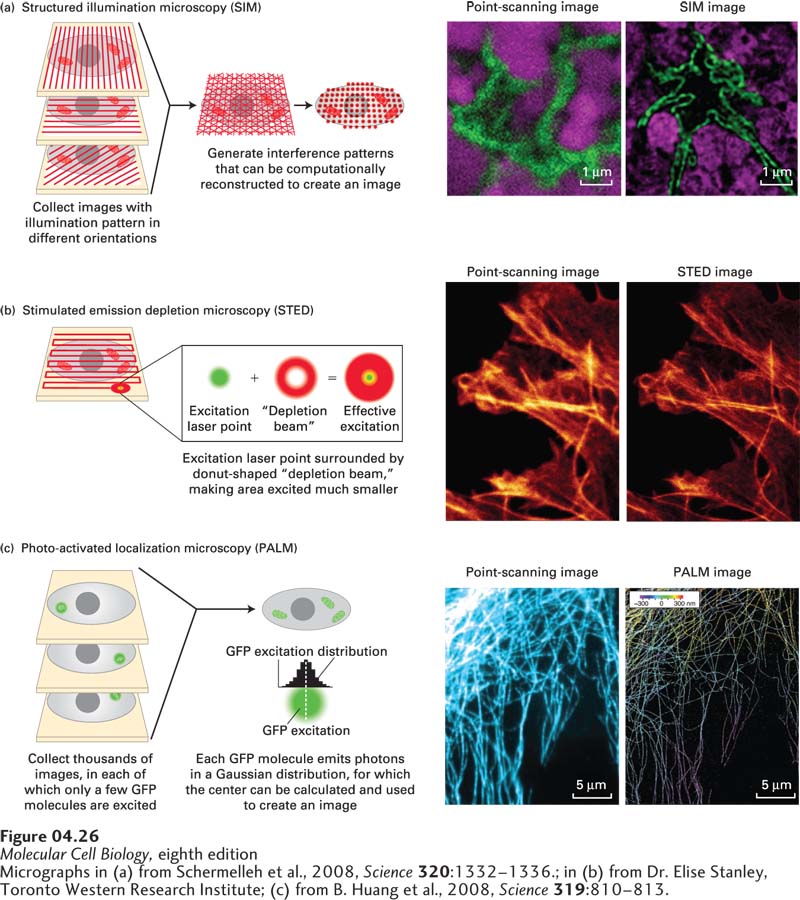
Super-Resolution Microscopy Can Localize Proteins to Nanometer Accuracy
As we discussed earlier, the theoretical resolution limit of the fluorescence microscope is about 0.2 µm (200 nm). Two new general approaches, collectively known as super-resolution microscopy, have been developed to get around this limitation. In the first type of approach, the illuminating light is patterned in such a way that higher-resolution images are obtained. In structured illumination microscopy (SIM), the specimen is illuminated with a pattern of light and dark stripes, and several images are taken as the illuminating pattern is rotated to generate Moiré fringes. Computational analysis of the images gives a resolution of about 100 nm, twice that of a conventional confocal microscope, as can be seen in a micrograph of part of the nuclear envelope (Figure 4-26a). SIM is especially good for live cell imaging, as three-dimensional images can be collected every 4 seconds. In stimulated emission depletion (STED) microscopy, the sample is scanned just as in point-scanning microscopy, but with an important difference: the focused excitation laser point is surrounded by a donut-shaped “depletion beam,” which effectively makes the area excited much smaller. Since the computer records the precise position of the spot excited and can record the emission from it, it can build up an impressive image, optimally yielding a resolution of 30 nm, greatly enhancing the detail one can see, for example, in an image of actin fibers (Figure 4-26b).
Page 154
[Micrographs in (a) from Schermelleh et al., 2008, Science320:1332–1336.; in (b) from Dr. Elise Stanley, Toronto Western Research Institute; (c) from B. Huang et al., 2008, Science319:810–813.]
EXPERIMENTAL FIGURE 4-26Super-resolution microscopy can generate light-microscope images with up to nanometer resolution. The theoretical resolution of the light microscope can be circumvented by super-resolution microscopy. (a) In structured illumination microscopy (SIM), the sample is illuminated by a pattern of light and dark stripes and several images are taken as the illumination is rotated. This technique generates interference patterns that can be mathematically reconstructed to generate a higher-resolution image. The images on the right show the similar fields of the nucleus (lamin, green; DNA, magenta) imaged by conventional point-scanning confocal microscopy and by SIM, which improves resolution about twofold. (b) In stimulated emission depletion microscopy (STED), the sample is scanned as in point-scanning microscopy, but with a very small point of light, generated by an emission laser and confined by a donut-shaped stimulated emission depletion zone. The sample at the right shows part of a cell stained for actin fibers after imaging by point-scanning microscopy and by STED. (c) In photoactivated localization microscopy (PALM), use is made of a variant of GFP that can be photoactivated by a wavelength different from its excitation wavelength. When a small number of GFP molecules are activated, and then excited, each will emit thousands of photons that can be collected. This generates a Gaussian curve centered on the location of the emitting GFP; the center provides the location of the GFP to nanometer accuracy. This process is reiterated hundreds of times to excite other GFP molecules, and a high-resolution image emerges. At the right, a confocal image of microtubules is compared with a corresponding super-resolution image in which the three-dimensional arrangement of the microtubules is color coded.
[Micrographs in (a) from Schermelleh et al., 2008, Science320:1332–1336.; in (b) from Dr. Elise Stanley, Toronto Western Research Institute; (c) from B. Huang et al., 2008, Science319:810–813.]
Page 155
The second general approach uses single-molecule detection and localization. To understand how this works, consider two fluorescent spots separated by 75 nm. When you try to image them, they each generate a Gaussian distribution of fluorescence, which overlap so much that they look like one spot. However, if you could image each spot individually and find the center of each Gaussian curve, you could “beat” the resolution limit and detect the two spots 75 nm apart. One way to do this, called photoactivated localization microscopy (PALM), relies on the ability of a variant of GFP to be photoactivated; that is, it can become fluorescent only after being activated by a specific wavelength of light, different from its excitation wavelength. Consider what happens when we activate just one such GFP molecule. When we then excite the sample, the one activated GFP emits many hundreds of photons, giving rise to a Gaussian distribution (Figure 4-26c). Although analysis of each photon does not tell us precisely where the GFP is, the center of the distribution can tell us where the GFP is located with nanometer accuracy. If we now activate another GFP, we can localize it individually with the same precision. In PALM, a small percentage of GFPs are activated and each localized with high precision, and then another set is activated and localized, and as additional cycles of activation and localization are recorded, a high-resolution image emerges. For example, the three-dimensional distribution of microtubules can be seen with much greater clarity than with conventional fluorescence microscopy (see Figure 4-26c). These types of images can take significant time to generate, so their use on samples of live cells is so far restricted. Nevertheless, because of the tremendous benefits of super-resolution microscopy, enormous efforts are being made to improve its sensitivity, speed, and spatial resolution, and we can expect rapid progress in the development of these approaches.