Nucleotide Excision Repairs Chemical Adducts that Distort Normal DNA Shape
Cells use nucleotide excision repair to fix DNA regions containing chemically modified bases, often called chemical adducts, that distort the normal shape of DNA locally. A key to this type of repair is the ability of certain proteins to slide along the surface of a double-stranded DNA molecule looking for bulges or other irregularities in the shape of the double helix. For example, this mechanism repairs thymine-thymine dimers, a common type of chemical adduct caused by UV light (Figure 5-37); these dimers interfere with both replication and transcription of DNA. Figure 5-38 illustrates how the nucleotide excision-repair system repairs damaged DNA.
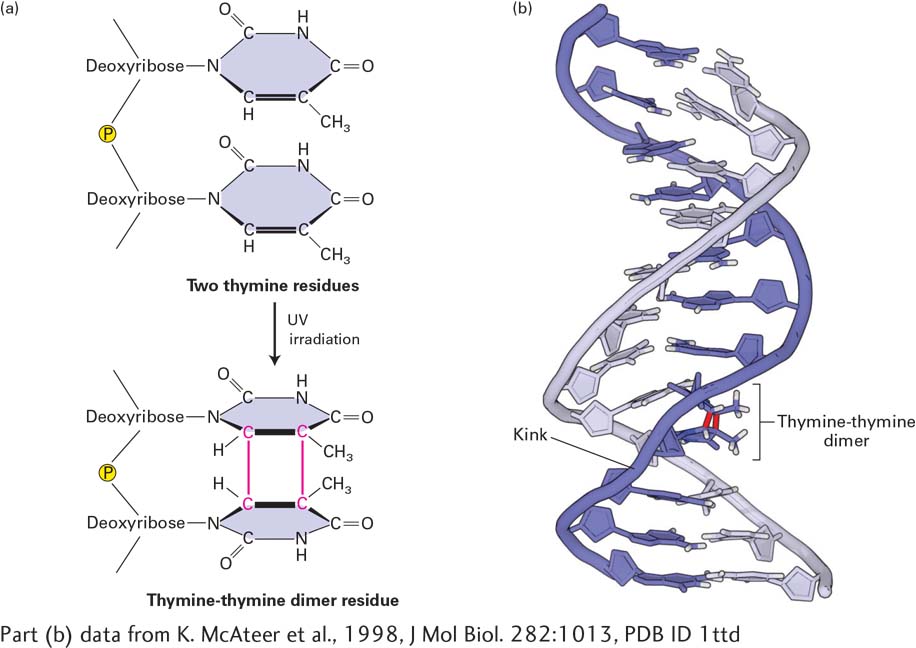
[Part (b) data from K. McAteer et al., 1998, J Mol Biol.282:1013, PDB ID 1ttd.]
FIGURE 5-37Formation of thymine-thymine dimers. (a) The most common type of DNA damage caused by UV irradiation is the formation of thymine-thymine dimers. (b) These lesions can be repaired by an excision-repair mechanism that recognizes the distortion they create in the shape of the DNA double helix. The red lines in (b) represent the UV-induced C—C bonds shown in (a).
[Part (b) data from K. McAteer et al., 1998, J Mol Biol.282:1013, PDB ID 1ttd.]
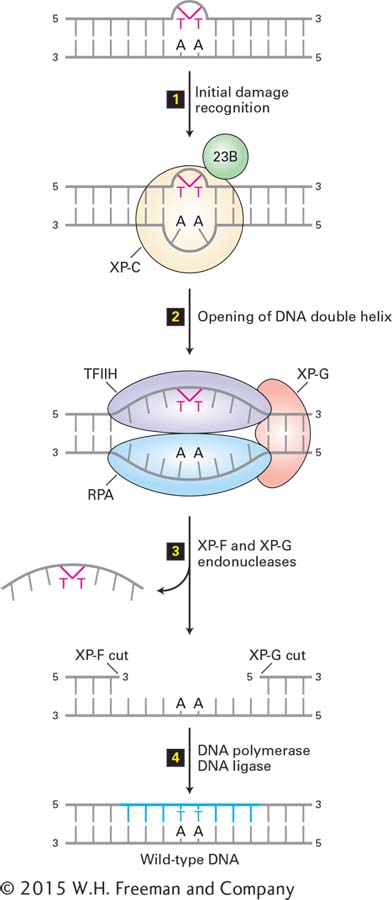
FIGURE 5-38Nucleotide excision repair in human cells. A DNA lesion that causes distortion of the double helix, such as a thymine-thymine dimer, is initially recognized by a complex of the XP-C (xeroderma pigmentosum C protein) and 23B proteins (step 1). This complex then recruits transcription factor TFIIH, whose helicase subunits, powered by ATP hydrolysis, partially unwind the double helix. XP-G and RPA proteins then bind to the complex and further unwind and stabilize the helix until a bubble of about 25 bases is formed (step 2). Then XP-G (now acting as an endonuclease) and XP-F, a second endonuclease, cut the damaged strand at points 24–32 bases apart on each side of the lesion (step 3). This releases the DNA fragment with the damaged bases, which is degraded to mononucleotides. Finally the gap is filled by DNA polymerase exactly as in DNA replication, and the remaining nick is sealed by DNA ligase (step 4). See J. Hoeijmakers, 2001, Nature411:366, and O. Schärer, 2003, Angewandte Chemie42:2946.
Page 207
Some 30 proteins are involved in the nucleotide excision-repair process, the first of which were identified through a study of the defects in DNA repair in cultured cells from individuals with xeroderma pigmentosum, a hereditary disease associated with a predisposition to cancer. Individuals with this disease frequently develop skin cancers called melanomas and squamous cell carcinomas if their skin is exposed to the UV rays in sunlight. The cells of affected patients lack a functional nucleotide excision-repair system. Mutations in any of at least seven different genes, called XP-A through XP-G, lead to inactivation of this repair system and cause xeroderma pigmentosum; all produce the same phenotype and have the same consequences. The functions of most of these XP proteins in nucleotide excision repair are now well understood (see Fig. 5-38).
Remarkably, five polypeptide subunits of TFIIH, a general transcription factor required for transcription of all genes (see Figure 9-19), are also required for nucleotide excision repair in eukaryotic cells. Two of these subunits have homology to helicases, as shown in Figure 5-38. In transcription, the helicase activity of TFIIH helps to unwind the DNA helix at the start site, allowing RNA polymerase to initiate the process (see Figure 9-19). It appears that nature has used a similar protein assembly in two different cellular processes that require helicase activity.
The use of shared subunits in transcription and in DNA repair may help explain the observation that DNA damage in higher eukaryotes is repaired at a much faster rate in regions of the genome being actively transcribed than in nontranscribed regions—a phenomenon called transcription-coupled repair. Since only a small fraction of the genome is transcribed in any one cell, transcription-coupled repair efficiently directs repair efforts to the most critical regions. In this system, if an RNA polymerase becomes stalled at a lesion on DNA (e.g., a thymine-thymine dimer), a complex of proteins CSA and CSB is recruited to the RNA polymerase, where they trigger opening of the DNA helix, recruitment of TFIIH, and the reactions of steps 2 through 4 depicted in Figure 5-38. CSA and CSB are named after the rare inherited developmental disorder called Cockayne syndrome that results from mutations in these proteins.