Cloned DNA Molecules Can Be Sequenced Rapidly by Methods Based on PCR
The complete characterization of any cloned DNA fragment requires determination of its nucleotide sequence. The technology used to determine the sequence of a DNA segment represents one of the most rapidly developing fields in molecular biology. In the 1970s, F. Sanger and his colleagues developed the chain-termination procedure, which served as the basis for most DNA sequencing methods for the next 30 years. The idea behind this method is to synthesize from the DNA fragment to be sequenced a set of daughter strands that are labeled at one end and terminate at one of the four nucleotides. Separation of the truncated daughter strands by gel electrophoresis, which can resolve strands that differ in length by a single nucleotide, can then reveal the lengths of all strands ending in G, A, T, or C. From these collections of strands of different lengths, the nucleotide sequence of the original DNA fragment can be established. The Sanger method has undergone many refinements and can now be fully automated, but because each DNA fragment requires a separate individual sequencing reaction, the overall rate at which new DNA sequences can be produced by this method is limited by the total number of reactions that can be performed at one time.
Page 244
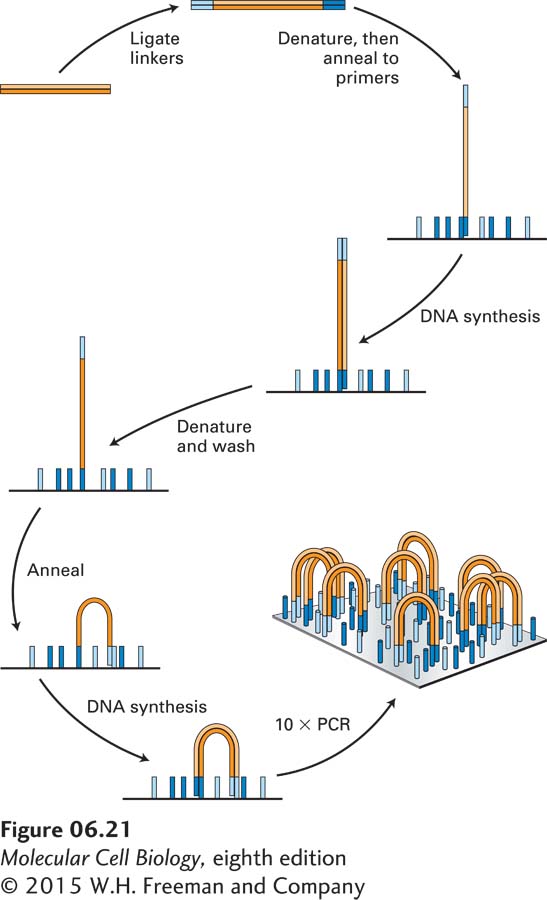
A breakthrough in sequencing technology occurred when methods were devised to allow a single sequencing instrument to carry out billons of sequencing reactions simultaneously by localizing them in tiny clusters on the surface of a solid substratum. In 2007, when these so-called next-generation sequencers became commercially available, the capacity for new sequence production increased enormously, and since then, because of improvements in the technology, it has been further increasing at an amazing pace—doubling every few months. In one popular sequencing method, billions of different DNA fragments to be sequenced are prepared by ligating double-stranded linkers to their ends (Figure 6-21). Next the DNA fragments are amplified by PCR using primers that match the linker sequences. This reaction differs from the standard PCR amplification shown in Figure 6-19 in that the primers used are covalently attached to a solid substratum. Thus, as the PCR amplification proceeds, one end of each daughter DNA strand is covalently linked to the substratum, and at the end of the amplification about a thousand identical PCR products are linked to the surface in in a tight cluster.
EXPERIMENTAL FIGURE 6-21Generation of clusters of identical DNA fragments attached to a solid support. A large collection of DNA fragments to be sequenced is ligated to double-stranded linkers, which become attached to each end of each fragment. The DNA is then amplified by PCR using primers matching the sequences of the linkers that are covalently attached to a solid substratum. Ten cycles of amplification yield about a thousand identical copies of each DNA fragment localized in a small cluster and attached at both ends to the solid substratum. These reactions are optimized to produce as many as 3 × 109 discrete, non-overlapping clusters that are ready to be sequenced.
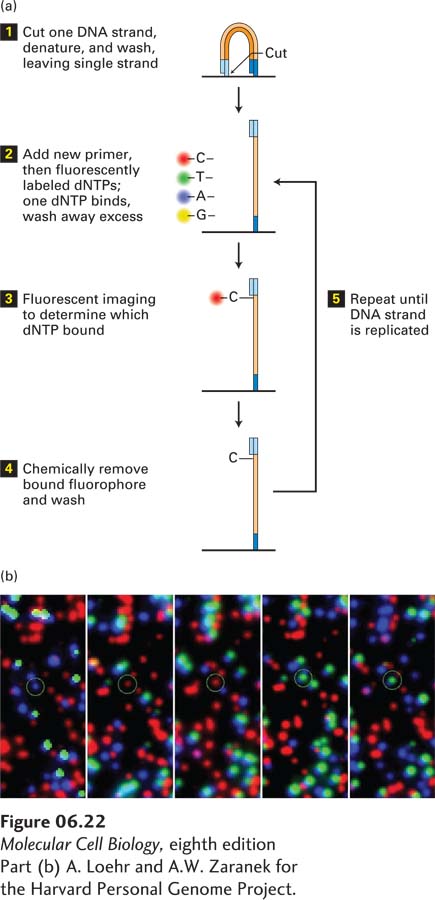
These clusters can then be sequenced by using a special microscope to image fluorescently labeled deoxyribonucleotide triphosphates (dNTPs) as they are incorporated by DNA polymerase one at a time into a growing DNA chain (Figure 6-22). First, one strand is cut and washed out, leaving a single-stranded DNA template. Then sequencing is carried out on the thousand or so identical templates in each cluster, one nucleotide at a time. All four dNTPs are fluorescently labeled and added to the sequencing reaction. After they are allowed to anneal, the substratum is imaged and the color of each cluster is recorded. Next the fluorescent tag is chemically removed and a new dNTP is allowed to bind. This cycle is repeated about a hundred times, resulting in billions of hundred-nucleotide-long sequences. The entire procedure may take about one day and yield some 1011 bases of sequence information.
[Part (b) A. Loehr and A.W. Zaranek for the Harvard Personal Genome Project.]
EXPERIMENTAL FIGURE 6-22Using fluorescent-tagged deoxyribonucleotide triphosphates for sequence determination. (a) The reaction begins with the cleaving of one strand of the amplified, clustered DNA (see Figure 6-21). After denaturation, a single DNA strand remains attached to the substratum. A synthetic oligodeoxynucleotide is used as the primer for the polymerization reaction, which also contains dNTPs, each fluorescently tagged with a different color. The fluorescent tag is designed to block the 3′ OH group on the dNTP so that once the fluorescent dNTP has been incorporated, further elongation is not possible. Because DNA polymerase will incorporate the same fluorescently labeled dNTP into each of the thousand or so identical DNA copies in a cluster, the entire cluster will be uniformly labeled with the same fluorescent color, which can be imaged in a special microscope. (b) Five images from the same field of view, each corresponding to an individual cycle of dNTP addition. Each colored dot represents a cluster of identical DNA fragments. After each image is made, the fluorescent tags are removed by a chemical reaction that leaves a new primer terminus available for the next cycle of dNTP addition. As can be seen for the circled colored dot, the color changes in each reaction cycle according to which nucleotide is added to the DNA fragments. A typical sequencing reaction may carry out a hundred polymerization cycles, allowing a hundred bases of sequence for each cluster to be determined. Thus a total sequencing reaction of this type may generate as much as 3 × 1011 bases of sequence information in about two days.
[Part (b) A. Loehr and A.W. Zaranek for the Harvard Personal Genome Project.]
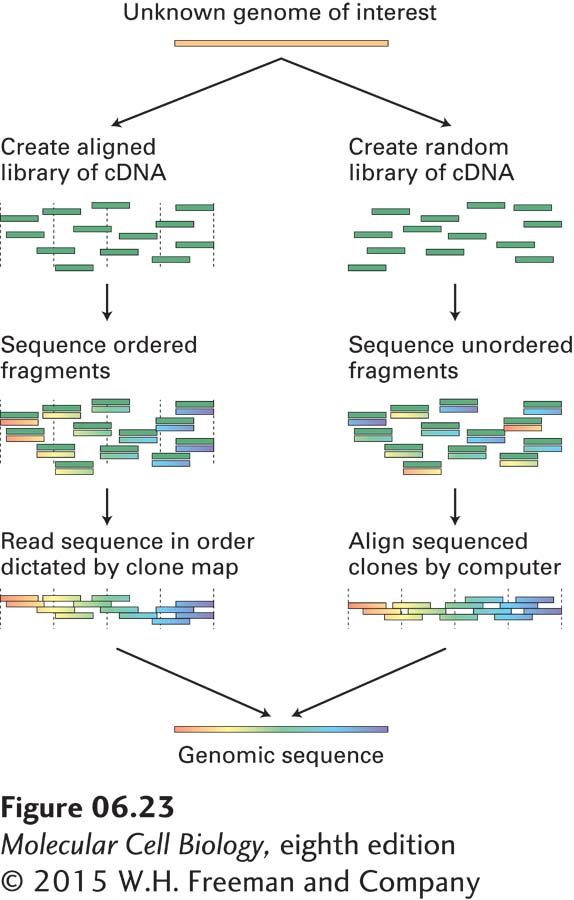
In order to sequence a long continuous region of genomic DNA, or even the entire genome of an organism, researchers usually employ one of the strategies outlined in Figure 6-23. The first method requires the isolation of a collection of cloned DNA fragments whose sequences overlap. Once the sequence of one of these fragments is determined, oligonucleotides based on that sequence can be chemically synthesized for use as primers in sequencing the adjacent overlapping fragments. In this way, the sequence of a long stretch of DNA is determined incrementally by sequencing of the overlapping cloned DNA fragments that compose it. A second method, which is called whole genome shotgun sequencing, bypasses the time-consuming step of isolating an ordered collection of DNA segments that span the genome. This method involves simply sequencing random clones from a genomic library. A sufficient number of clones are chosen for sequencing so that on average, each segment of the genome is sequenced about 10 times. This degree of coverage ensures that each segment of the genome is sequenced more than once. The entire genomic sequence is then assembled using a computer algorithm that aligns all the segments using their regions of overlap. Whole genome shotgun sequencing is the fastest and most cost-effective method for sequencing long stretches of DNA, and most genomes, including the human genome, have been sequenced by this method.
Page 245
EXPERIMENTAL FIGURE 6-23Two strategies for assembling whole genome sequences. One method (left) depends on isolating and assembling a set of cloned DNA fragments that span the genome. This can be done by matching cloned fragments by hybridization or by alignment of restriction-site maps. The DNA sequence of the ordered clones can then be assembled into a complete genomic sequence. The alternative method (right) depends on the relative ease of automated DNA sequencing and bypasses the laborious step of ordering a DNA library. By sequencing enough random clones from the library so that each segment of the genome is represented from 3 to 10 times, it is possible to reconstruct the genomic sequence by computer alignment of the very large number of sequence fragments.