Plasmid Expression Vectors Can Be Designed for Use in Animal Cells
Although bacterial expression systems can be used successfully to create large quantities of some proteins, bacteria cannot be used in all cases. Many experiments designed to examine the function of a protein in an appropriate cellular context require expression of a genetically modified protein in cultured animal cells. To accomplish this, genes are cloned into specialized eukaryotic expression vectors and are introduced into cultured animal cells by a process called transfection. Two common methods for transfecting animal cells differ in whether the recombinant vector DNA is or is not integrated into the host-cell genomic DNA.
In both methods, cultured animal cells must be treated to facilitate their initial uptake of the recombinant plasmid vector. This can be done by exposing cells to a preparation of lipids that penetrates the plasma membrane, increasing its permeability to DNA. Alternatively, subjecting cells to a brief electric shock of several thousand volts, a technique known as electroporation, makes them transiently permeable to DNA. Usually the plasmid DNA is added in sufficient concentration to ensure that a large proportion of the cultured cells will receive at least one copy. Researchers have also harnessed viruses for transfection; viruses can be modified to contain DNA of interest, which is then introduced into host cells by simply infecting them with the recombinant virus.
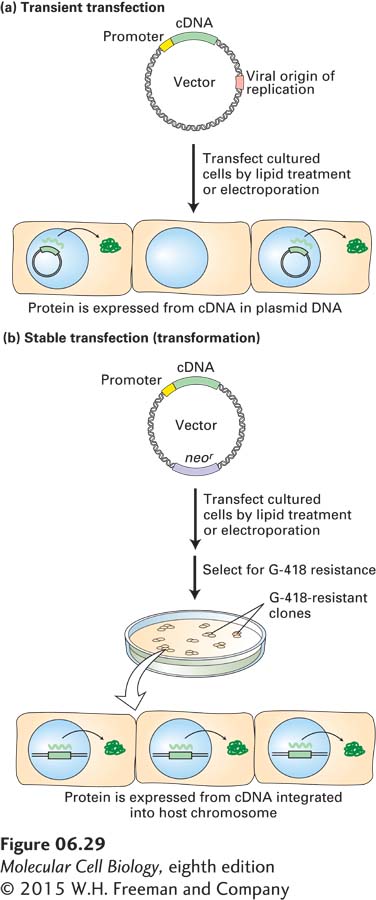
EXPERIMENTAL FIGURE 6-29Transient and stable transfection with specially designed plasmid vectors permits expression of cloned genes in cultured animal cells. Both methods employ plasmid vectors that contain the usual elements—replication origin, selectable marker (e.g., ampr), and polylinker—that permit propagation in E. coli as well as a cloned cDNA with an adjacent animal promoter. For simplicity, these elements are not depicted. (a) In transient transfection, the plasmid vector contains a replication origin from a virus that can replicate in the cultured animal cells. Since the vector is not incorporated into the genome of the cultured cells, production of the cDNA-encoded protein continues for only a limited time. (b) In stable transfection, the vector carries a selectable marker such as neor, which confers resistance to G-418. The relatively few transfected animal cells that integrate the exogenous DNA into their genomes are selected on medium containing G-418. Because the vector is integrated into the genome, these stably transfected, or transformed, cells will continue to produce the cDNA-encoded protein as long as the culture is maintained. See the text for discussion.
Transient Transfection The simpler of the two transfection methods, called transient transfection, employs a plasmid vector similar to the yeast shuttle vectors described previously. For use in mammalian cells, plasmid vectors are engineered to carry a replication origin derived from a virus that infects mammalian cells, a strong promoter recognized by mammalian RNA polymerase, and the cloned cDNA encoding the protein to be expressed adjacent to the promoter (Figure 6-29a). Once such a plasmid vector enters a mammalian cell, the viral replication origin allows it to replicate efficiently, generating numerous plasmids from which the protein is expressed. However, during cell division, such plasmids are not faithfully segregated into both daughter cells, and in time, a substantial fraction of the cells in a culture will not contain a plasmid, hence the name transient transfection.
Page 252
Stable Transfection (Transformation) If an introduced vector integrates into the genome of the host cell, that genome is permanently altered, and the cell is said to be transformed. Integration is most likely accomplished by endogenous enzymes that normally function in DNA repair and recombination. A commonly used selectable marker is the gene for neomycin phosphotransferase (designated neor), which confers resistance to a toxic compound chemically related to neomycin, known as G-418. The basic procedure for expressing a cloned cDNA by stable transfection is outlined in Figure 6-29b. Only those cells that have integrated the expression vector into the host chromosome will survive and give rise to a clone in the presence of a high concentration of G-418. Because integration occurs at random sites in the genome, individual transformed G-418-resistant clones will differ in their rates of transcribing the inserted cDNA. Therefore, the stable transfectants are usually screened to identify those that produce the protein of interest at the highest levels.
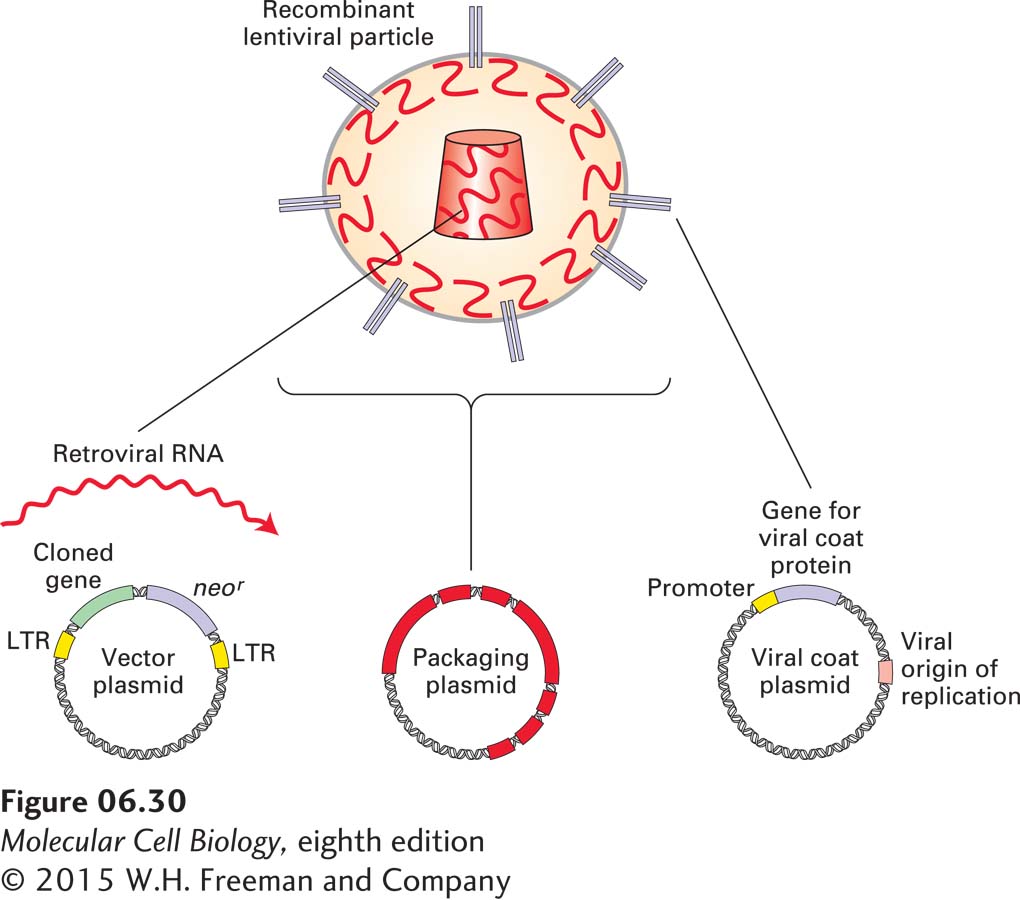
Retroviral Expression Systems Researchers have exploited the basic mechanisms used by viruses for introduction of their genetic material into animal cells and its subsequent insertion into chromosomal DNA to greatly increase the efficiency by which a modified gene can be stably expressed in animal cells. One such viral expression system is derived from a class of retroviruses known as lentiviruses. As shown in Figure 6-30, three different plasmids, introduced into cells by transient transfection, are used to produce recombinant lentivirus particles suitable for efficient introduction of a cloned gene into target animal cells. The first plasmid, known as the vector plasmid, contains a cloned gene of interest next to a selectable marker such as neor flanked by lentivirus LTR sequences. The left LTR sequence directs synthesis of an RNA molecule that carries lentiviral LTR sequences at either end and thus has many of the properties of native retroviral RNA. In an appropriate host, this LTR-bearing RNA can be packaged into viral particles and then introduced into a target cell by viral infection. In the target cell, the LTR sequences direct the copying of the RNA into double-stranded DNA by reverse transcription and the integration of that DNA into chromosomal DNA following a sequence of events depicted in Figure 8-14. A second plasmid, known as the packaging plasmid, carries all the viral genes except for the major viral envelope protein, necessary for packaging LTR-containing viral RNA into a functional lentivirus particle. The final plasmid allows expression of a viral envelope protein that, when incorporated into a recombinant lentivirus, allows the resulting hybrid virus particles to infect a desired target cell type. A common envelope protein used in this context is the glycoprotein of the vesicular stomatitis virus (VSV-G protein), which can readily replace the normal lentivirus envelope protein on the surface of completed virus particles and allows the resulting virus particles to infect a wide variety of mammalian cell types, including hematopoietic stem cells, neurons, and muscle and liver cells. After cell infection, the cloned gene flanked by the viral LTR sequences is reverse-transcribed into DNA, which is transported into the nucleus and then integrated into the host genome. If necessary, as in the case of stable transfection, cells with a stably integrated cloned gene and neor marker can be selected by resistance to G-418. Many of the techniques for inactivating the function of specific genes (see Section 6.5) require that an entire population of cultured cells be genetically modified simultaneously. Engineered lentiviruses are particularly useful for such experiments because they infect cells with such high efficiency that every cell in a population will receive at least one copy of the lentivirus-borne plasmid.
See text for discussion.
EXPERIMENTAL FIGURE 6-30Retroviral vectors can be used for efficient integration of cloned genes into the mammalian genome.
See text for discussion.
Page 253
[Courtesy Ashish Maurya.]
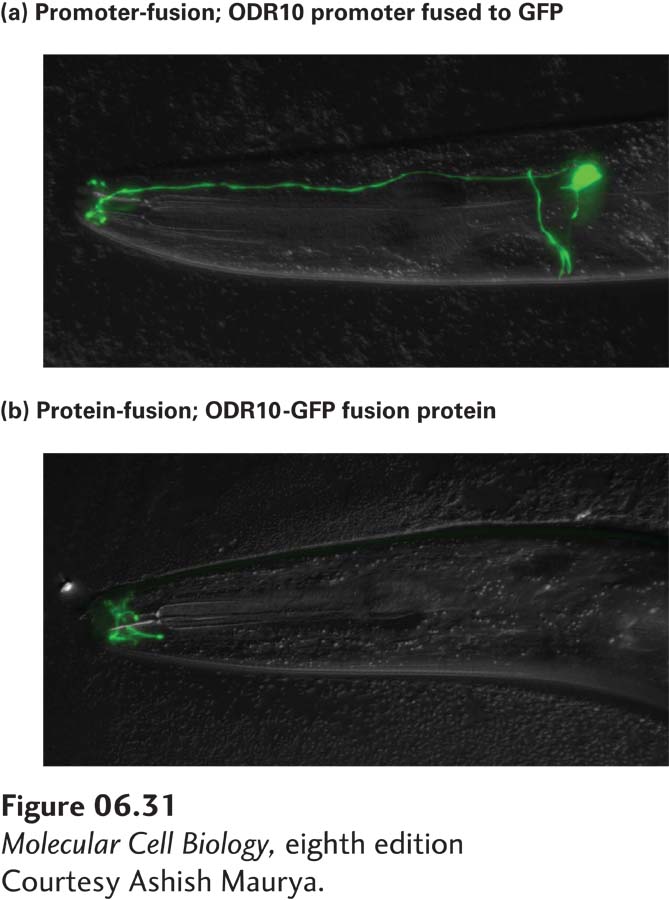
EXPERIMENTAL FIGURE 6-31Gene and protein tagging facilitate cellular localization of proteins expressed from cloned genes. In this experiment, the gene encoding a chemical odorant receptor, Odr10, of C. elegans was fused to the gene encoding green fluorescent protein (GFP). (a) A promoter-fusion was generated by linking just the promoter of Odr10 to the coding sequence for GFP. The result is that GFP is expressed in the cytoplasm of the same specific sensory neurons in the head of C. elegans where Odr10 is expressed. (b) A protein-fusion was constructed by linking GFP to the end of the full-length Odr10 coding sequence. In this case, the Odr10-GFP fusion protein is targeted to the membrane at the tip of the sensory neurons and is apparent only at the distal end of the sensory cilia. The observed distribution can be inferred to reflect the normal location of Odr10 protein in specific neurons. Because the promoter-fusion shown in (a) lacks the Odr10 localization sequences, the expressed GFP fills the entire cell cytoplasm rather than being localized just to the distal tip of the sensory cilia.
[Courtesy Ashish Maurya.]
Gene and Protein Tagging Expression vectors can provide a way to study the expression and intracellular localization of eukaryotic proteins. Such studies often rely on the use of a reporter protein, such as green fluorescent protein (GFP), that can be conveniently detected in cells (see Figure 4-16). Here we describe two ways to create a hybrid gene that connects expression of the reporter protein to that of the protein of interest. When the hybrid gene is reintroduced into cells, either by transfection with a plasmid expression vector containing the modified gene or by creation of a transgenic animal as described in Section 6.5, the expression of the reporter protein can be used to determine where and when a gene is expressed. This method provides data similar to that from the in situ hybridization experiments described previously, but often with greater resolution and sensitivity.
Figure 6-31 illustrates the use of two different types of GFP-tagging experiments to study the expression of an odorant receptor protein in C. elegans. When the promoter for the odorant receptor is linked directly to the coding sequence of GFP in a configuration usually known as a promoter-fusion, GFP is expressed in specific neurons, filling the cytoplasm of those neurons. In contrast, when the hybrid gene is constructed by linking GFP to the coding sequence of the odorant receptor, the resulting protein-fusion can be localized by GFP fluorescence at the distal cilia in sensory neurons, the site at which the receptor protein is normally located.
An alternative to GFP tagging for detecting the intracellular location of a protein is to modify the gene of interest by appending to it a short DNA sequence that encodes a short stretch of amino acids recognized by a known monoclonal antibody. Such a short peptide that can be bound by an antibody is called an epitope; hence this method is known as epitope tagging. After transfection of cells with a plasmid expression vector containing the modified gene, the expressed epitope-tagged form of the protein can be detected by immunofluorescence labeling of the cells with the monoclonal antibody specific for the epitope. The choice of whether to use a short epitope or GFP to tag a given protein often depends on what types of modification a cloned gene can tolerate and still remain functional.