Linkage Studies Can Map Disease Genes with a Resolution of About 1 Centimorgan
Without going into all the technical considerations, let’s see how the allele conferring a particular dominant disease trait (e.g., familial hypercholesterolemia) might be mapped. The first step is to obtain DNA samples from all the members of a family containing individuals that exhibit the disease. The DNA from each affected and unaffected individual is then analyzed to determine that individual’s genotype for a large number of known DNA polymorphisms (either STR or SNP markers can be used). The segregation pattern of each DNA polymorphism within the family is then compared with the segregation pattern of the disease under study. Polymorphisms that are not linked to the disease allele will not show any significant tendency to co-
In practice, segregation data are usually collected from different families exhibiting the same disease and pooled. The more families that can be examined, the greater the statistical significance of any evidence for linkage that can be obtained, and the greater the precision with which the distance between a linked DNA polymorphism and a disease allele can be measured. Most family studies have a maximum of about 100 individuals in whom linkage between a disease gene and a panel of DNA polymorphisms can be tested. This number of individuals sets the practical upper limit on the resolution of such a mapping study to about 1 centimorgan, or a physical distance of about 7.5 × 105 bp.
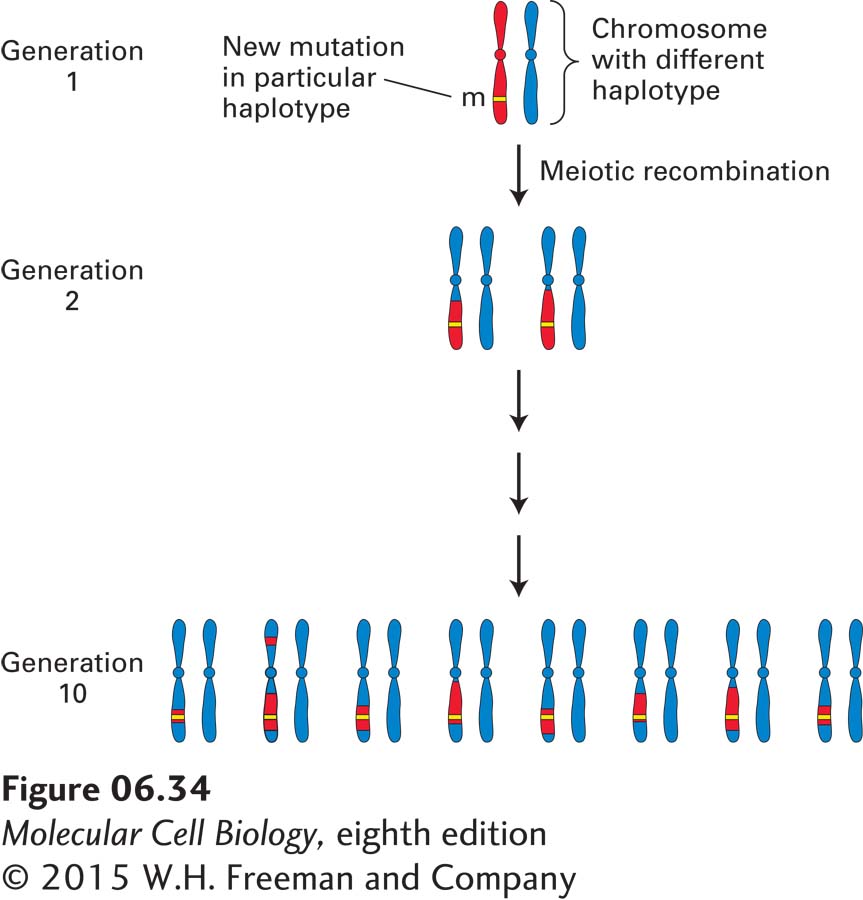
A phenomenon called linkage disequilibrium is the basis for an alternative mapping strategy, which can often afford a higher degree of resolution. This approach can be applied to a genetic disease commonly found in a particular population that results from a single mutation that occurred many generations in the past. The DNA polymorphisms carried by the ancestral chromosome in which the mutation occurred are collectively known as the haplotype of that chromosome. As the disease allele is passed from one generation to the next, only the polymorphisms that are closest to the disease gene will not be separated from it by recombination. After many generations, the region that contains the disease gene will be evident because it will be the only region of the chromosome that will carry the haplotype of the ancestral chromosome conserved through many generations (Figure 6-34). By assessing the distribution of specific markers in all affected individuals in a population, geneticists can identify DNA markers tightly associated with the disease, thus localizing the disease-
Page 257