Further Analysis Is Needed to Locate a Disease Gene in Cloned DNA
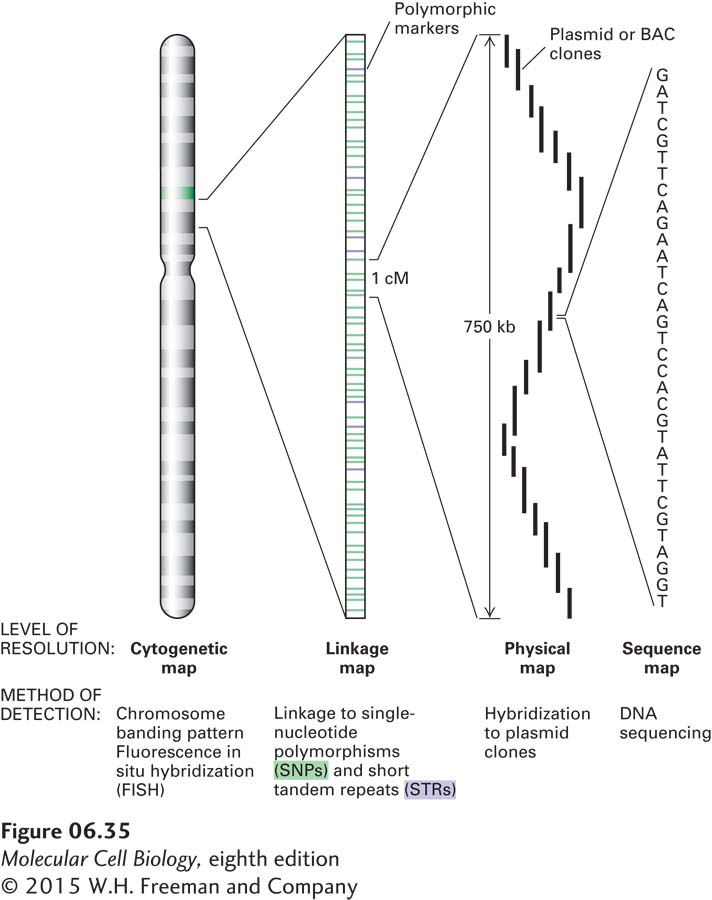
Although linkage mapping can usually locate a human disease gene to a region containing about 105 bp, as many as 10 different genes may be located in a region of this size. The ultimate objective of a mapping study is to locate the gene of interest within a cloned segment of DNA and then to determine the nucleotide sequence of this fragment. The relative scales of a chromosomal genetic map and physical maps corresponding to ordered sets of plasmid clones and the nucleotide sequence are shown in Figure 6-35.
FIGURE 6-35Relationship between genetic and physical maps of a human chromosome. The diagram depicts a human chromosome analyzed at different levels of detail. The chromosome as a whole can be viewed in the light microscope when it is in a condensed state that occurs at metaphase, and the approximate location of specific sequences can be determined by fluorescence in situ hybridization (FISH). At the next level of detail, genetic traits can be mapped relative to DNA-based genetic markers. Local segments of the chromosome can be analyzed at the level of DNA sequences identified by Southern blotting or PCR. Finally, important genetic differences can be most precisely defined by differences in the nucleotide sequence of the chromosomal DNA.
One strategy for further localizing a disease gene within the genome is to identify mRNA encoded by DNA in the region under study. Comparison of gene expression in tissues from normal and affected individuals may suggest tissues in which a particular disease gene is normally expressed. For instance, a mutation that phenotypically affects muscle, but no other tissue, might be in a gene that is expressed only in muscle tissue. The expression of mRNA in both normal and affected individuals is generally determined by Northern blotting, microarray analysis, or in situ hybridization of labeled DNA or RNA to tissue sections. Northern blots, in situ hybridization, or microarray experiments permit comparison of both the level of expression and the sizes of mRNAs in mutant and wild-type tissues. Although the sensitivity of in situ hybridization is lower than that of Northern blot analysis, it can be very helpful in identifying an mRNA that is expressed at low levels in a given tissue but at very high levels in a subclass of cells within that tissue. An mRNA that is altered or missing in various individuals affected with a disease compared with wild-type individuals would be an excellent candidate for encoding the protein whose disrupted function causes that disease.
In many cases, point mutations that give rise to disease-causing alleles result in no detectable change in the level of expression or electrophoretic mobility of mRNAs. Thus, if comparison of the mRNAs expressed in normal and affected individuals reveals no detectable differences in the candidate mRNAs, a search for point mutations in the DNA regions encoding the mRNAs is undertaken. Now that highly efficient methods for sequencing DNA are available, researchers frequently determine the sequences of candidate regions of DNA isolated from affected individuals to identify point mutations. The overall strategy is to search for a coding sequence that consistently shows possibly deleterious alterations in DNA from individuals that exhibit the disease. A limitation of this approach is that the region near the affected gene may carry naturally occurring polymorphisms unrelated to the gene of interest. Such polymorphisms, not functionally related to the disease, can lead to misidentification of the DNA fragment carrying the gene of interest. For this reason, the more mutant alleles available for analysis, the more likely that a gene will be correctly identified.