Engineered CRISPR–Cas9 Systems Allow Precise Genome Editing
A recently developed technique allows precise alterations to be made in genomic DNA sequences by a process known as genome editing. This method is based on a natural mechanism that evolved to protect bacterial cells against foreign DNA such as phage DNA. This mechanism, called CRISPR (clustered regularly interspaced short palindromic repeats), is named after the curious arrays of tandem repeated sequences found in about half of the bacterial genomes that have been sequenced. The arrays of repeated sequences are flanked by a conserved set of genes, known as Cas (CRISPR-associated) genes, that show similarity to genes encoding nucleases.
The breakthrough in understanding the function of CRISPR sequences came from the observation that one set of repeated sequence elements in the array often matched short segments in phage genomes. It was subsequently shown that the CRISPR element and associated Cas genes confer on bacterial cells the ability to cleave phage DNA at precisely the site that corresponds to repeated sequences in the CRISPR array. The acquisition of immunity to a phage by means of CRISPR takes place in two stages. In the first stage, a phage-infected bacterium carrying a CRISPR system cleaves the phage DNA into short segments and adds those segments to the CRISPR array so that they are interspersed with highly conserved repeats. In the second stage, transcription of the CRISPR array and processing of the resulting RNA yields mature bipartite RNA molecules that carry both the conserved repeat sequences and a phage-derived spacer sequence. The repeat sequence assembles with a second RNA molecule called tracrRNA that provides a scaffold for binding to Cas proteins to form an interference complex, and the phage-derived sequence guides the complex to a specific target sequence through base pairing. Once targeted to a specific DNA sequence, nucleases in the Cas proteins cleave both strands of the target DNA molecule at a site adjacent to the region base-paired with the guide sequence.
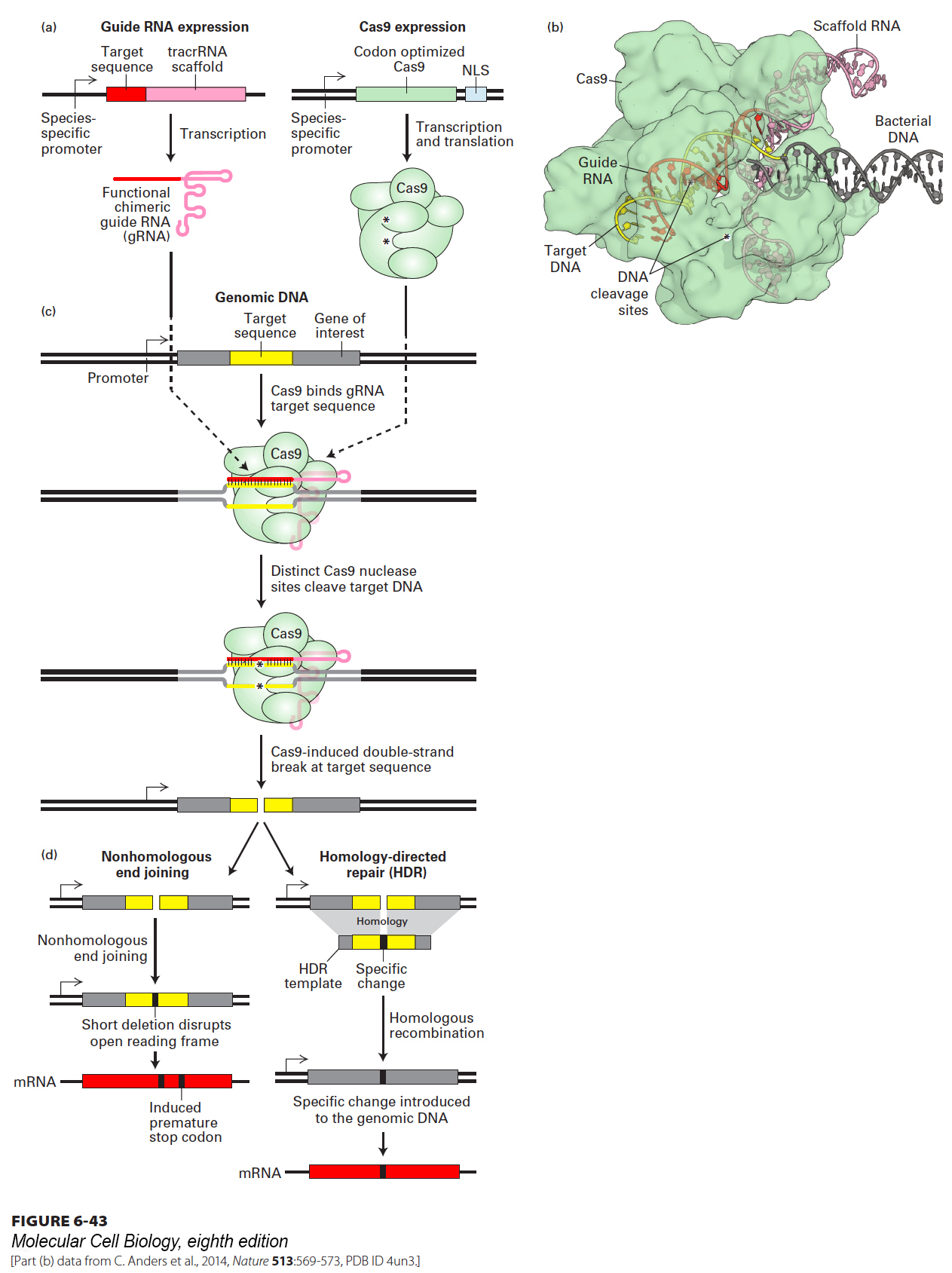
Although CRISPR elements have been found only in prokaryotic organisms, researchers proposed that the guide RNA and Cas proteins should be able to perform their functions if expressed in eukaryotic cells. To adapt CRISPR to function in virtually any cell type, minimal systems consisting of the nuclease Cas9 and an engineered guide RNA have been developed (Figure 6-43a). Cas9 contains all the enzymatic activities necessary for genome editing, including two separate endonuclease activities—one for each strand of DNA. The guide RNA is composed of two regions: the first is made up of two complementary sequences that form a double-stranded hairpin structure designed to bind to Cas9, and the second region of about 20 nt provides the targeting activity and is designed to perfectly match a specific site in genomic DNA. When both Cas9 and the guide RNA are expressed in a recipient cell, Cas9 will cleave both DNA strands at the chromosomal site specified by the guide RNA sequence (Figure 6-43b,c). Transfection with expression plasmids for Cas9 and a specific guide RNA has been shown to give rise to specific DNA cleavages in cells derived from a variety of organisms, including Drosophila, C. elegans, zebrafish, mouse, rat, and human. A particularly efficient way to modify the germ line of an organism begins with microinjecting Cas9 mRNA and a guide RNA into a mouse zygote. The resulting double-strand break in the specific target sequence is typically repaired by a set of enzymes that ligate the free DNA ends back together in a process known as nonhomologous end joining (Figure 6-43d). Typically, a few base pairs are deleted at the site of cleavage because nucleases can remove bases from the free DNA ends before the end joining reaction is completed. If the cleavage site is within a gene coding sequence, the deletion of several bases will usually cause a frameshift mutation, thus inactivating the targeted gene.
Page 267
[Part (b) data from C. Anders et al., 2014, Nature513:569-573, PDB ID 4un3.]
FIGURE 6-43Single-nucleotide mutations can be introduced into the genome using an engineered CRISPR-Cas9 system. (a) The genome of a target cell can be modified by expression of the double-stranded DNA endonuclease Cas9 and a guide RNA. Expression of these components can be achieved by transfection with plasmids carrying genes for Cas9 and the guide RNA or by direct injection of Cas9 mRNA and guide RNA. The guide RNA is composed of two parts: a sequence that folds into a hairpin scaffold structure that binds to Cas9, and a sequence of approximately 20 nt corresponding to the targeted site in the genome. Expression of these components can be achieved by transfection with plasmids carrying genes for Cas9 and the guide RNA or by direct injection of Cas9 mRNA and guide RNA. (b) A complex of guide RNA bound to Cas9 is targeted to the genome by base pairing of the guide RNA with the complementary genomic DNA sequence. This structure allows the two distinct nuclease active sites of Cas9 to cleave both strands of the target DNA adjacent to the heteroduplex formed with the guide RNA. (c) By this mechanism, the expression of both Cas9 and a bipartite guide RNA designed to target a specific gene sequence leads to a double-strand cleavage of the target gene. (d) Cleaved DNA can be repaired via a nonhomologous end joining (NHEJ) process, which usually removes a small number of bases at the cleavage site. If the cleavage occurs in a coding sequence, NHEJ will usually inactivate gene function by producing a frameshift mutation. If a ∼100-nt single-stranded DNA segment that spans the sequences flanking the cleavage site is injected along with Cas9 mRNA and the guide RNA, the cleaved DNA can be repaired by homologous recombination (homology-directed repair, HDR). By this mechanism, single base changes can be introduced into the repaired genomic DNA.
[Part (b) data from C. Anders et al., 2014, Nature513:569-573, PDB ID 4un3.]
An important refinement of this method of genome editing is the addition of a segment of DNA, typically about 100 nt long, that matches the sequences flanking the cleavage site. When homologous DNA is present, the free ends can be repaired by homologous recombination using the added DNA segment (see Figure 6-43d). By appropriate design of the guide RNA to specify the cleavage site, and by introduction of appropriate sequence alterations in the added homologous DNA segment, exact single-base changes in the target DNA can be produced. In a dramatic demonstration of this genome editing method, a single-base mutation in Crygc, a gene for gamma-crystallin, that leads to the formation of cataracts was corrected back to the wild-type form by injection of a mutant mouse zygote with Cas9 mRNA, a guide RNA that directs cleavage at the site of the mutation, and a 90-nt DNA segment spanning the cleavage site and containing the wild-type Crygc sequence (see the chapter-opening photo). This experiment also illustrates the remarkable selectivity of CRISPR-Cas9 targeting, since correction of the mutant allele requires that the guide RNA discriminate between the mutant and wild-type alleles, which differ by only one base pair.
Page 268
In Section 6.4, we saw that human genetic mapping and association studies have identified a large number of genetic variants, many representing single-nucleotide polymorphisms that predispose people to one or another inherited diseases. The ability to use CRISPR-Cas9 systems to produce specific single-base changes in the mammalian germ line now opens the possibility of creating the equivalent alleles in a well-controlled experimental organism such as the mouse.