Stored Sequences Suggest Functions of Newly Identified Genes and Proteins
As discussed in Chapter 3, proteins with similar functions often contain similar amino acid sequences that correspond to important functional domains in the three-dimensional structure of the proteins. By comparing the amino acid sequence of the protein encoded by a newly cloned gene with the sequences of proteins of known function, an investigator can look for sequence similarities that provide clues to the function of the encoded protein. Because of the degeneracy in the genetic code, related proteins invariably exhibit more sequence similarity than the genes encoding them. For this reason, protein sequences, rather than the corresponding DNA sequences, are usually compared.
The most widely used computer program for this purpose is known as BLAST (basic local alignment search tool). The BLAST algorithm divides the “new” protein sequence (known as the query sequence) into shorter segments and then searches the database for significant matches to any of the stored sequences. The matching program assigns a high score to identically matched amino acids and a lower score to matches between amino acids that are related (e.g., hydrophobic, polar, positively charged, negatively charged) but not identical. When a significant match is found for a segment, the BLAST algorithm searches locally to extend the region of similarity. After searching is completed, the program ranks the matches between the query protein and various known proteins according to their p-values. This parameter is a measure of the probability of finding such a degree of similarity between two protein sequences by chance. The lower the p-value, the greater the sequence similarity between two sequences. A p-value less than about 10−3 is usually considered significant evidence that two proteins share a common ancestor. Many alternative computer programs have been developed that can detect relationships between proteins that are more distantly related to each other than can be detected by BLAST. The development of such methods is currently an active area of bioinformatics research.
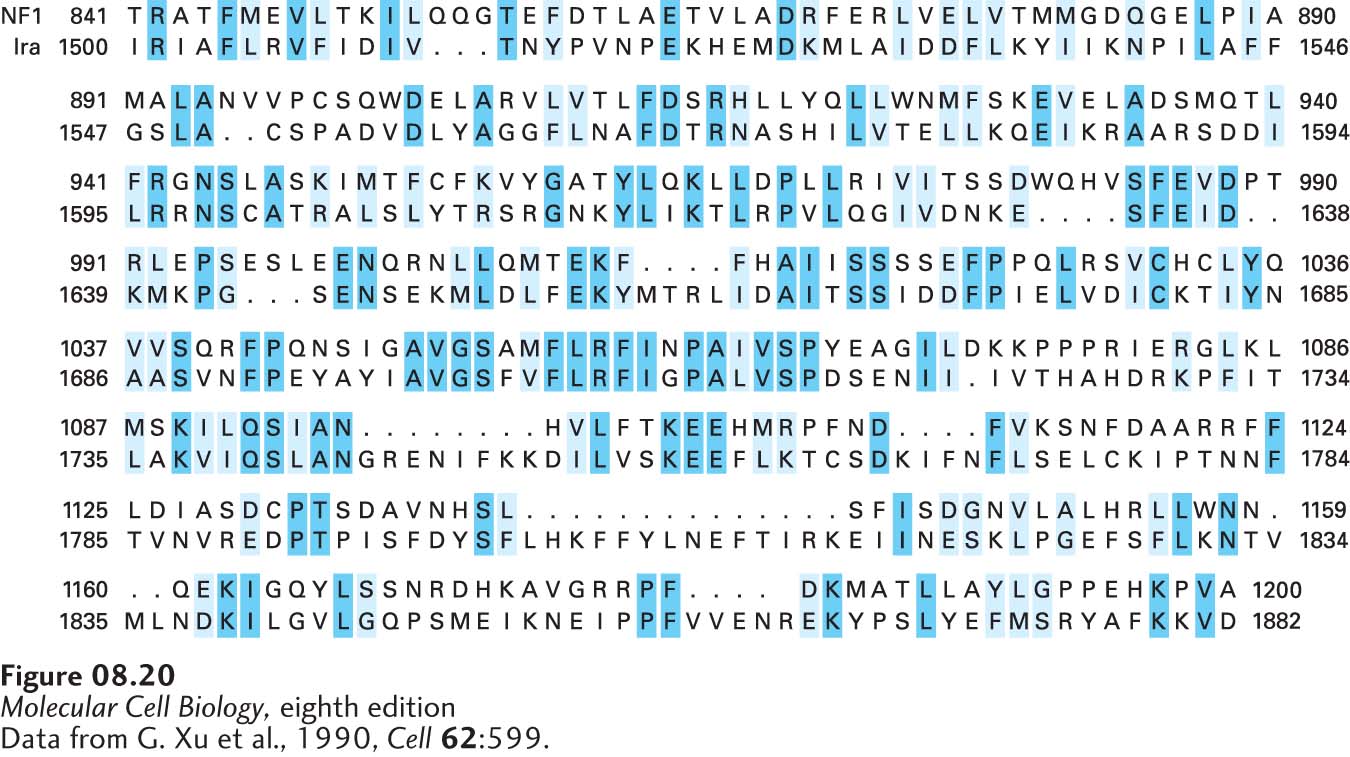
To illustrate the power of this sequence comparison approach, let’s consider the human gene NF1. Mutations in NF1 are associated with the inherited disease neurofibromatosis 1, in which multiple tumors develop in the peripheral nervous system, causing large protuberances in the skin. After a cDNA clone of NF1 was isolated and sequenced, the deduced sequence of the NF1 protein was checked against all other protein sequences in GenBank. A region of NF1 protein was discovered to have considerable homology to a portion of the yeast protein called Ira (Figure 8-20). Previous studies had shown that Ira is a GTPase-activating protein (GAP) that modulates the GTPase activity of the monomeric G protein called Ras (see Figure 3-34). As we examine in detail in Chapter 16, GAP and Ras proteins normally function to control cell replication and differentiation in response to signals from neighboring cells. Functional studies on the normal NF1 protein, obtained by expression of the cloned wild-type gene, showed that it did, indeed, regulate Ras activity, as suggested by its homology with Ira. These findings suggest that patients with neurofibromatosis express a mutant NF1 protein in cells of the peripheral nervous system, leading to abnormally high signaling through RAS protein leading to excessive cell division and formation of the tumors characteristic of the disease.
[Data from G. Xu et al., 1990, Cell62:599.]
FIGURE 8-20Comparison of the regions of human NF1 protein and S. cerevisiae Ira protein that show significant sequence similarity. The NF1 and the Ira sequences are shown on the top and bottom lines of each row, respectively, in the one-letter amino acid code (see Figure 2-14). Amino acids that are identical in the two proteins are highlighted in dark blue. Amino acids with chemically similar but nonidentical side chains are highlighted in light blue. Black dots indicate “gaps” in the upper and lower protein sequences, inserted in order to maximize the alignment of homologous amino acids. The BLAST p-value for these two sequences is 10−28, indicating a high degree of similarity.
[Data from G. Xu et al., 1990, Cell62:599.]
Page 325
Even when the BLAST algorithm finds no significant similarities, a query sequence may nevertheless share a short sequence with known proteins that is functionally important. Such short segments recurring in many different proteins, referred to as structural motifs, generally have similar functions. Several such motifs are described in Chapter 3 and illustrated in Figure 3-10. To search for these and other motifs in a new protein, researchers compare the query protein sequence with a database of known motif sequences.