Genome annotation identifies various types of sequence.
Genomes contain many different types of sequence, among them protein-coding genes. Protein-coding genes are themselves composed of different regions, including regulatory elements that specify when and where an RNA transcript is produced, noncoding introns that are removed from the RNA transcript during RNA processing (Chapter 3), and protein-coding exons that contain the codons that specify the amino acid sequence of a polypeptide chain (Chapter 4). Genomes also contain coding sequences for RNAs that are not translated into protein (noncoding RNAs), such as ribosomal RNA, transfer RNA, and other types of small RNA molecule (Chapter 3). Finally, while much of the DNA in the genomes of multicellular organisms is transcribed at least in some cell types, the functions of a large portion of these transcripts in metabolism, physiology, development, or behavior are unknown.
Genome annotation is the process by which researchers identify the various types of sequence present in genomes. Genome annotation is essentially an exercise in adding commentary to a genome sequence that identifies which types of sequence are present and where they are located. It can be thought of as a form of pattern recognition, where the patterns are regularities in sequence that are characteristic of protein-coding genes or other types of sequence.
An example of genome annotation is shown in Fig. 13.3. Genes present in one copy per genome are indicated in orange. Most single-copy genes are protein-coding genes. In some computer programs that analyze genomes, the annotation of a single-copy gene also specifies any nearby regulatory regions that control transcription, the intron–exon boundaries in the gene, and any known or predicted alternative forms in which the introns and exons are spliced (Chapter 3). Each single-copy gene is given a unique name and its protein product identified. Note in Fig. 13.3 that single-copy genes can differ in size from one gene to the next. The annotations in Fig. 13.3 also specify the locations of sequences that encode RNAs that are not translated into proteins, as well as various types of repeated sequence. Most genomes also contain some genes that are present in multiple copies that originate from gene duplication; however these are usually identifiable in the same way as single-copy genes because the copies often differ enough in sequence that they can be distinguished.
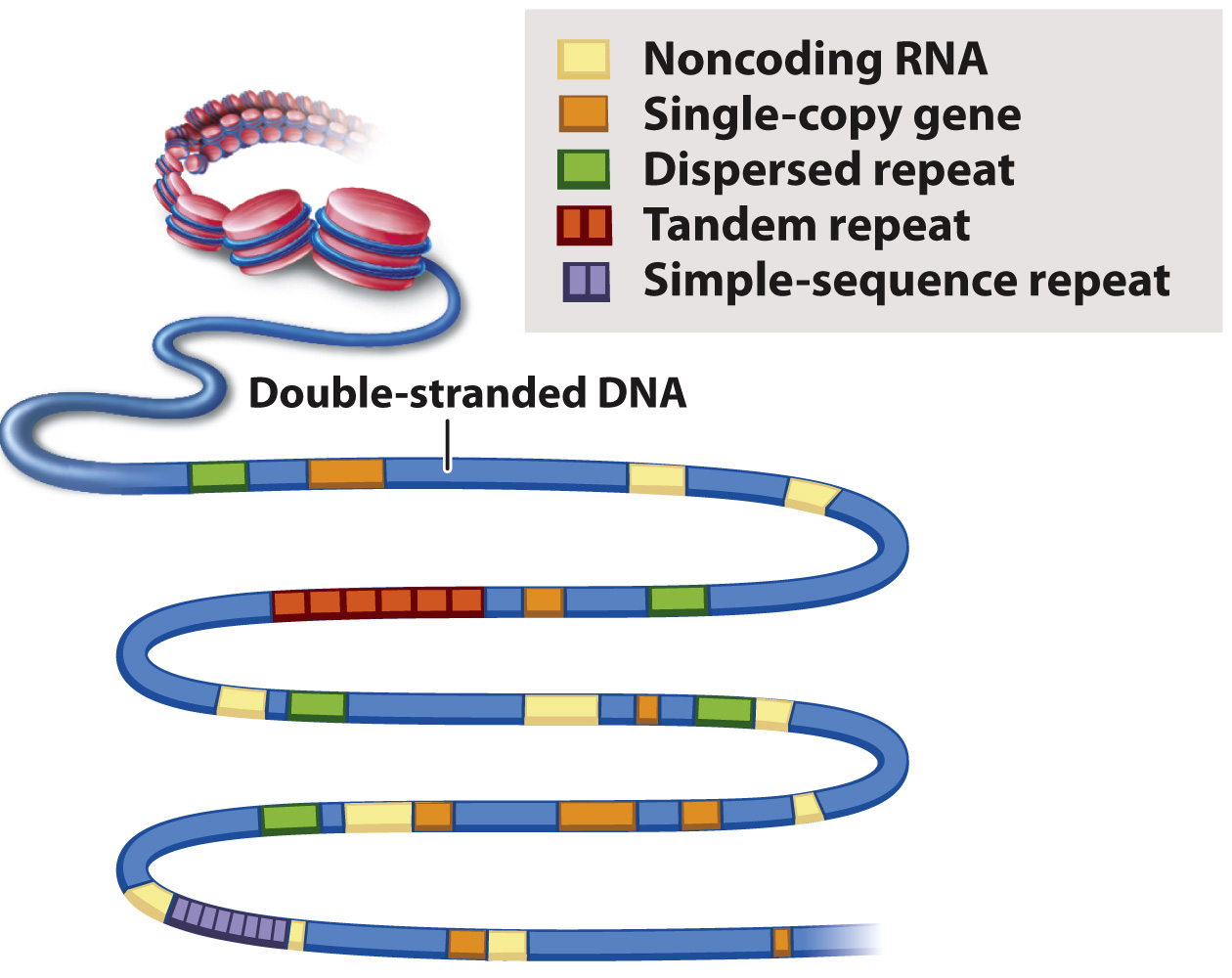
FIG. 13.3 Genome annotation. Given the DNA sequence of a genome, researchers can pinpoint locations of various types of sequence.
Small genomes such as that of HIV and other viruses can be annotated by hand, but for large genomes like the human genome, computers are essential. In the human genome, some protein-coding genes extend for more than a million nucleotides. Roughly speaking, if the sequence of the approximately 3 billion nucleotides in a human egg or sperm was printed in normal-sized type like this, the length of the ribbon would stretch 4000 miles (6440 kilometers), about the distance from Fairbanks, Alaska, to Miami, Florida. By contrast, for the approximately 10,000 nucleotides in the HIV genome, the ribbon would extend a mere 70 feet (21 meters).
Genome annotation is an ongoing process because, as macromolecules and their functions and interactions become better understood, the annotations to the genome must be updated. A sequence that is annotated as nonfunctional today may be found to have a function tomorrow. For this reason, the annotation of certain genomes—including the human genome—will certainly continue to change.