Secondary structures result from hydrogen bonding in the polypeptide backbone.
Hydrogen bonds can form between the carbonyl group in one peptide bond and the amide group in another, thus allowing localized regions of the polypeptide chain to fold. This localized folding is a major contributor to the secondary structure of the protein. In the early 1950s, American structural biologists Linus Pauling and Robert Corey used X-ray crystallography to study the structure of proteins. This technique was pioneered by British biochemists Dorothy Crowfoot Hodgkin, Max Perutz, and John Kendrew, among others (Fig. 4.5). Pauling and Corey studied crystals of highly purified proteins and discovered that two types of secondary structure are found in many different proteins. These are the α (alpha) helix and the β (beta) sheet. Both these secondary structures are stabilized by hydrogen bonding along the polypeptide backbone.
Page 74
HOW DO WE KNOW?
What are the shapes of proteins?
BACKGROUND The three-dimensional shapes of proteins can be determined by X-ray crystallography. One of the pioneers in this field was Dorothy Crowfoot Hodgkin, who used this technique to define the structures of cholesterol, vitamin B12, penicillin, and insulin. She was awarded the Nobel Prize in Chemistry in 1964 for her early work. Max Perutz and John Kendrew shared the Nobel Prize in Chemistry in 1962 for defining the structures of myoglobin and hemoglobin using this method.
METHOD X-ray crystallography can be used to determine the shape of proteins, as well as other types of molecules. The first step, which can be challenging, is to make a crystal of the protein molecules. A crystal is a solid structure in which the atoms of a protein (or any other molecule) are in an ordered and repeating pattern in three dimensions. Then X-rays are aimed at the crystal while it is rotated. Some X-rays pass through the crystal, while others are scattered in different directions when they hit atoms of the proteins. A film or other detector records the pattern as a series of spots, which is known as a diffraction pattern. The locations and intensities of these spots can be used to infer the position and arrangement of the atoms in the molecule.
RESULTS The X-ray diffraction pattern for hemoglobin looks like this:

FIG. 4.5
FOLLOW-UP WORK Linus Pauling and Robert Corey used X-ray crystallography to determine two types of secondary structures commonly found in proteins—the α helix and the β sheet. Today, this technique is a common method for determining the shape of proteins.
SOURCES Crowfoot, D. 1935. “X-Ray Single Crystal Photographs of Insulin.” Nature 135:591–592; Kendrew, J. C., et al. 1958. “A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis.” Nature 181:662–666.
In α helices, like the one shown in Fig. 4.6, the polypeptide backbone is twisted tightly in a right-handed coil with 3.6 amino acids per complete turn. The helix is stabilized by hydrogen bonds that form between each amino acid’s carbonyl group (C=O) and the amide group (N–H) four residues ahead in the sequence, as indicated by the dashed lines in Fig. 4.6. Note that the R groups project outward from the α helix. The chemical properties of the projecting R groups largely determine where the α helix is positioned in the folded protein, and how it might interact with other molecules.
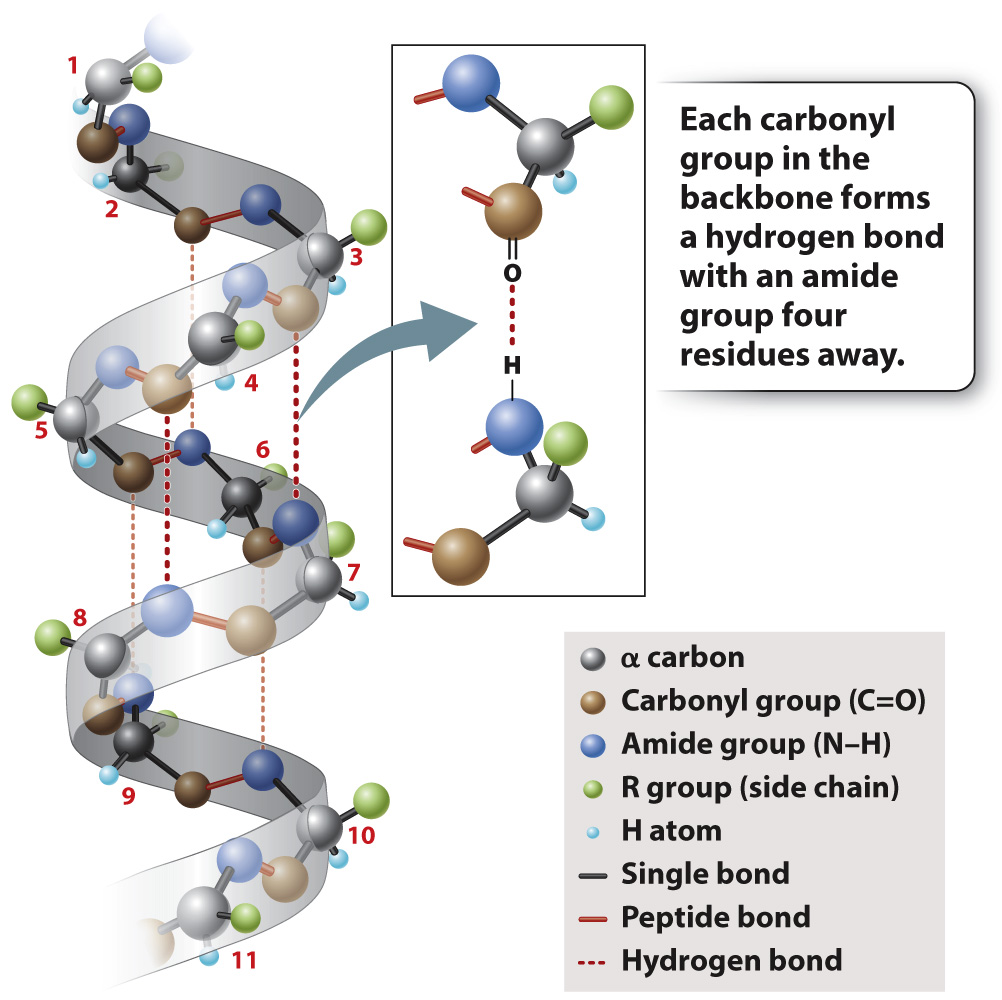
FIG. 4.6 An α helix. Hydrogen bonding between carbonyl and amide groups in the backbone stabilizes the helix.
The other secondary structure that Pauling and Corey found is the β sheet, depicted in Fig. 4.7. In a β sheet, the polypeptide folds back and forth on itself, forming a pleated sheet that is stabilized by hydrogen bonds between carbonyl groups in one chain and amide groups in the other chain across the way (dashed lines). The R groups project alternately above and below the plane of the β sheet. β sheets typically consist of 4 to 10 polypeptide chains aligned side by side, with the amides in each chain hydrogen-bonded to the carbonyls on either side (except for those at the ends of each strand).
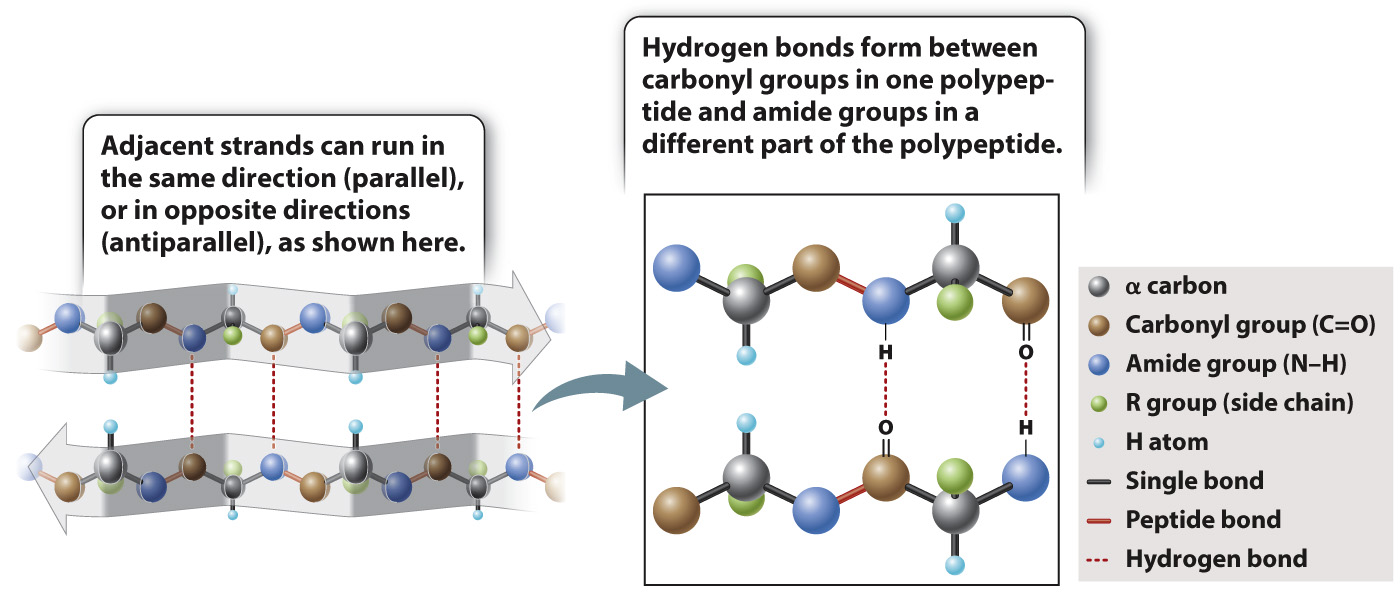
FIG. 4.7 A β sheet. Hydrogen bonds between neighboring strands stabilize the structure.
β sheets are typically denoted by broad arrows, where the direction of the arrow runs from the amino end of the polypeptide segment to the carboxyl end. In Fig. 4.7, the arrows run in opposite directions, and the polypeptide chains are said to be antiparallel. β sheets can also be formed by hydrogen bonding between polypeptide chains that are parallel (pointing in the same direction). However, the antiparallel configuration is more stable because the carbonyl and amide groups are more favorably aligned for hydrogen bonding.
Page 75