17.2 Changes in Chromatin Structure Affect the Expression of Genes
One type of gene control in eukaryotic cells is accomplished through the modification of chromatin structure. In the nucleus, histone proteins associate to form octamers, around which helical DNA tightly coils to create chromatin (see Figure 11.4). In a general sense, this chromatin structure represses gene expression. For a gene to be transcribed, transcription factors, other regulator proteins, and RNA polymerase must bind to the DNA. How can these events take place with DNA wrapped tightly around histone proteins? The answer is that, before transcription, chromatin structure changes and the DNA becomes more accessible to the transcriptional machinery.
DNase I Hypersensitivity
Several types of changes are observed in chromatin structure when genes become transcriptionally active. As genes become transcriptionally active, regions around the genes become highly sensitive to the action of DNase I (see Chapter 11). These regions, called DNase I hypersensitive sites, frequently develop about 1000 nucleotides upstream of the start site of transcription, suggesting that the chromatin in these regions adopts a more open configuration during transcription. This relaxation of the chromatin structure allows regulatory proteins access to binding sites on the DNA. Indeed, many DNase I hypersensitive sites correspond to known binding sites for regulatory proteins. At least three different processes affect gene regulation by altering chromatin structure: (1) chromatin remodeling; (2) the modification of histone proteins; and (3) DNA methylation. Each of these mechanisms will be discussed in the sections that follow.
CONCEPTS
Sensitivity to DNase I digestion indicates that transcribed DNA assumes an open configuration before transcription.
Chromatin Remodeling
Some transcription factors and other regulatory proteins alter chromatin structure without altering the chemical structure of the histones directly. These proteins are called chromatin-remodeling complexes. They bind directly to particular sites on DNA and reposition the nucleosomes, allowing transcription factors and RNA polymerase to bind to promoters and initiate transcription (Figure 17.1).
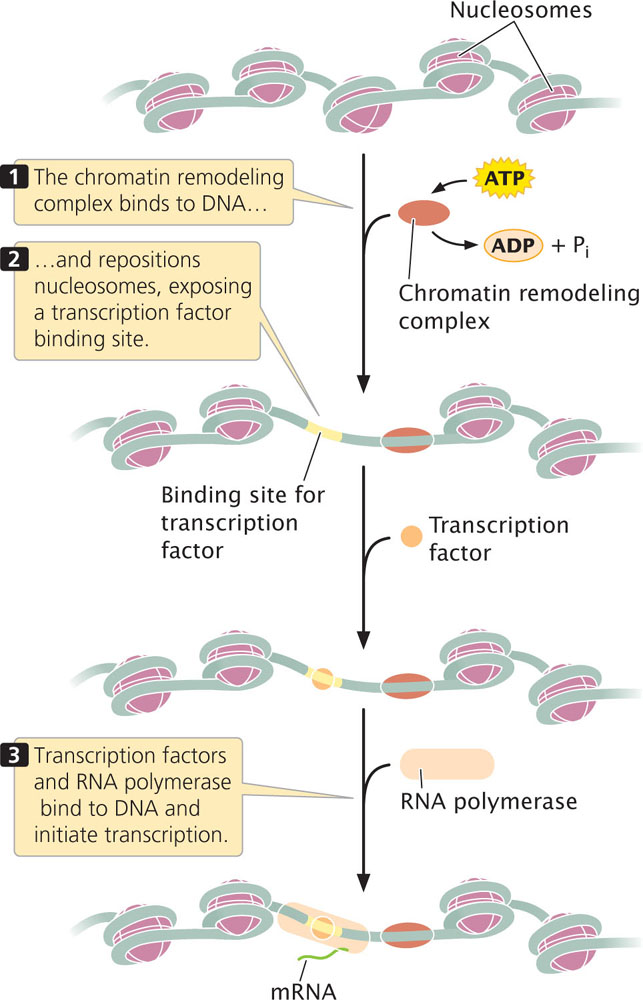
One of the best-studied examples of a chromatin-remodeling complex is SWI–SNF, which is found in yeast, humans, Drosophila, and other organisms. This complex utilizes energy derived from the hydrolysis of ATP to reposition nucleosomes, exposing promoters in the DNA to the action of other regulatory proteins and RNA polymerase.
Evidence suggests at least two mechanisms by which remodeling complexes reposition nucleosomes. First, some remodeling complexes cause the nucleosome to slide along the DNA, allowing DNA that was wrapped around the nucleosome to occupy a position in between nucleosomes, where it is more accessible to proteins affecting gene expression (see Figure 17.1). Second, some complexes cause conformational changes in the DNA, in nucleosomes, or in both so that DNA that is bound to the nucleosome assumes a more exposed configuration.
Chromatin-remodeling complexes are targeted to specific DNA sequences by transcriptional activators or repressors that attach to a remodeling complex and then bind to the promoters of specific genes. There is also evidence that chromatin-remodeling complexes work together with enzymes that alter histones, such as acetyltransferase enzymes (see next section), to change chromatin structure and expose DNA for transcription.
Histone Modification
Histones in the octamer core of the nucleosome have two domains: (1) a globular domain that associates with other histones and the DNA and (2) a positively charged tail domain that interacts with the negatively charged phosphate groups on the backbone of DNA. The tails of histone proteins are often modified by the addition or removal of phosphate groups, methyl groups, or acetyl groups. Another modification of histones is ubiquitination, in which small molecules called ubiquitin are added or removed from the histones. All of these modifications have sometimes been called the histone code, because they encode information that affects how genes are expressed. The histone code affects gene expression by altering chromatin structure directly or, in some cases, by serving as recognition sites for proteins that bind to DNA and that then regulate transcription.
Methylation of histones
One type of histone modification is the addition of methyl groups to the tails of histone proteins. These modifications can bring about either the activation or the repression of transcription, depending on which particular amino acids in the histone tail are methylated. Enzymes called histone methyltransferases add methyl groups to specific amino acids (usually lysine or arginine) of histone proteins. Other enzymes, called histone demethylases, remove methyl groups from histones. Many of the enzymes and proteins that modify histones, such as methyltransferases and demethylases, do not bind to specific DNA sequences, and must be recruited to specific chromatin sites. Sequence-specific binding proteins, pre-existing histone modifications, and RNA molecules serve to recruit histone-modifying enzymes to specific sites.
A common modification is the addition of three methyl groups to lysine 4 in the tail of the H3 histone protein, abbreviated H3K4me3 (K is the abbreviation for lysine). Histones containing the H3K4me3 modification are frequently found near promoters of transcriptionally active genes in eukaryotes. Studies have identified proteins that recognize and bind to H3K4me3, including nucleosome remodeling factor (NURF). NURF and other proteins that recognize H3K4me3 have a common protein-binding domain that binds to the H3 histone tail and then alters chromatin packing, allowing transcription to take place. Research has also demonstrated that some transcription factors, which are necessary for the initiation of transcription (see Chapter 13 and Section 17.3), directly bind to H3K4me3.
Acetylation of Histones
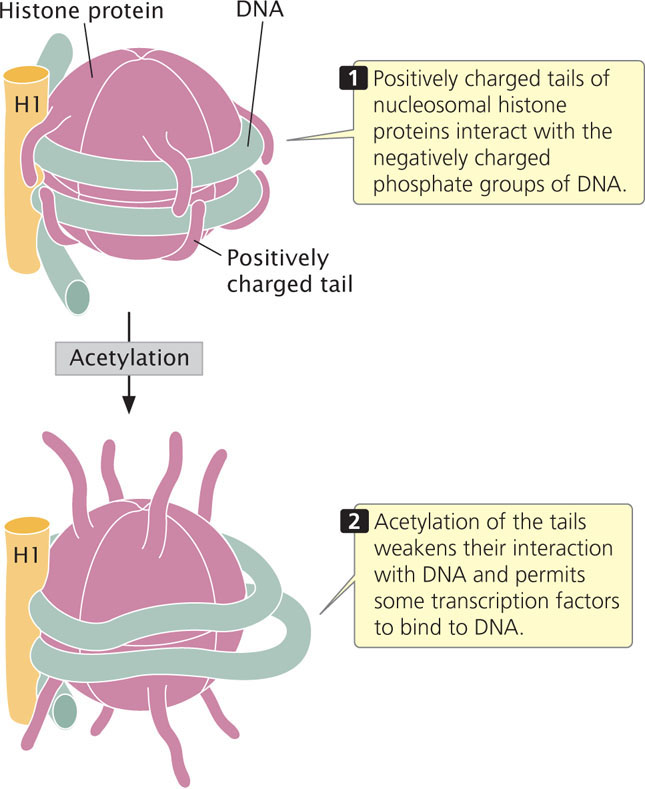
Another type of histone modification that affects chromatin structure is acetylation, the addition of acetyl groups (CH3CO) to histone. The acetylation of histones usually stimulates transcription. For example, the addition of a single acetyl group to lysine 16 in the tail of the H4 histone prevents the formation of the 30-nm chromatin fiber (see Figure 11.4), causing the chromatin to be in an open configuration and available for transcription. In general, acetyl groups destabilize chromatin structure, allowing transcription to take place (Figure 17.2). Acetyl groups are added to histone proteins by acetyltransferase enzymes; other enzymes called deacetylases strip acetyl groups from histones and restore chromatin structure, which represses transcription. Certain transcription factors (see Chapter 13) and other proteins that regulate transcription either have acetyltransferase activity or attract acetyltransferases to DNA.
The Acetylation of Histones Controls Flowering in Arabidopsis
The importance of histone acetylation in gene regulation is demonstrated by the control of flowering in Arabidopsis thaliana, a plant with a number of characteristics that make it an excellent genetic model for plant systems (see further information on it in the Reference Guide to Model Genetic Organisms at the end of the book). The time at which flowering takes place is critical to the life of a plant; if flowering is initiated at the wrong time of year, pollinators may not be available to fertilize the flowers or environmental conditions may be unsuitable for the survival and germination of the seeds. Consequently, flowering time in most plants is carefully regulated in response to multiple internal and external cues, such as plant size, photoperiod, and temperature.
Among the many genes that control flowering in Arabidopsis is flowering locus C (FLC), which plays an important role in suppressing flowering until after an extended period of coldness (a process called vernalization). The FLC gene encodes a regulator protein that represses the activity of other genes that affect flowering (Figure 17.3). As long as FLC is active, flowering remains suppressed. The activity of FLC is controlled by another locus called flowering locus D (FLD), the key role of which is to stimulate flowering by repressing the action of FLC. In essense, flowering is stimulated because FLD represses the repressor. How does FLD repress FLC?
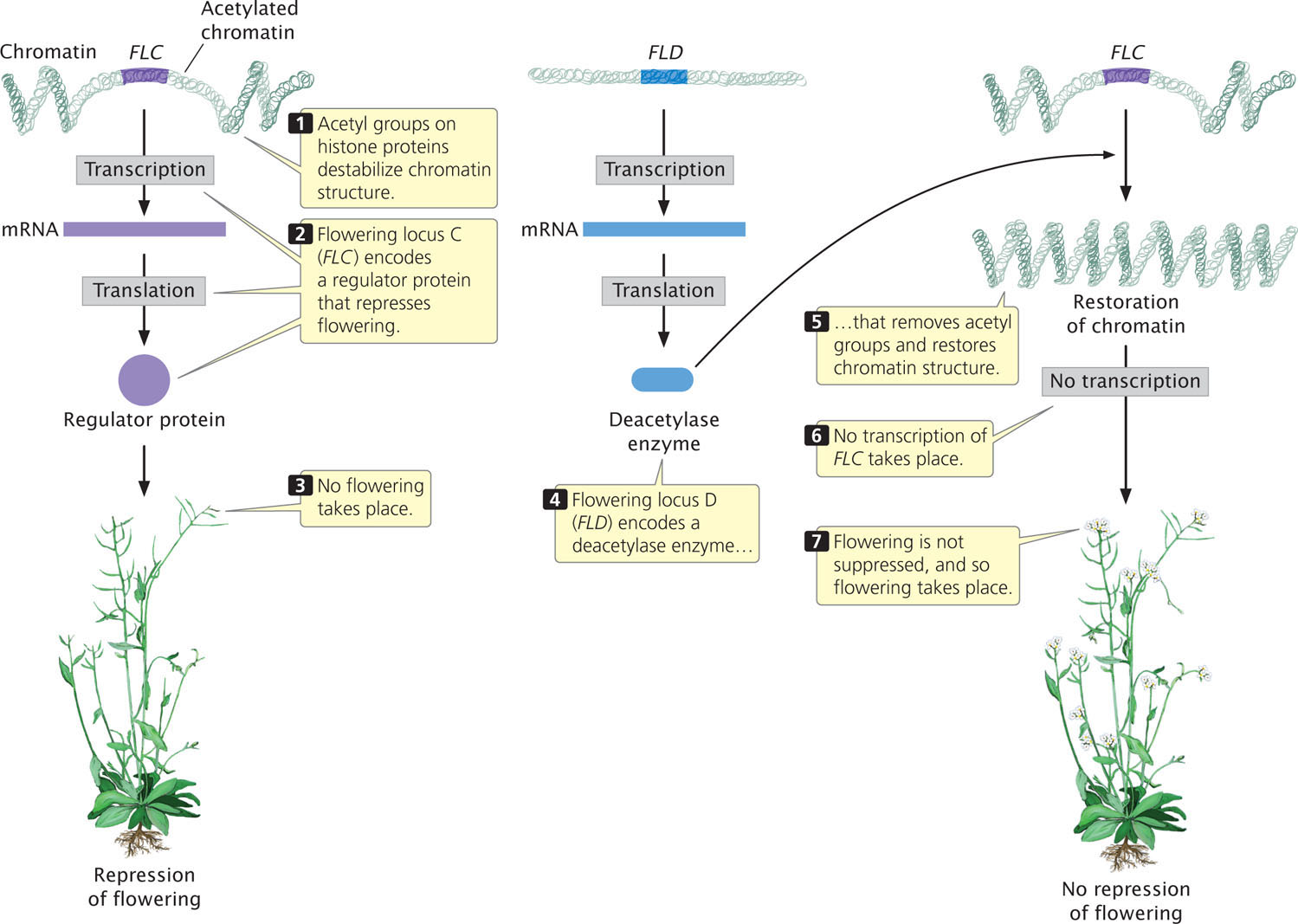
FLD encodes a deacetylase enzyme, which removes acetyl groups from histone proteins in the chromatin surrounding FLC (see Figure 17.3). The removal of acetyl groups from histones alters chromatin structure and inhibits transcription. The inhibition of transcription prevents FLC from being transcribed and removes its repression on flowering. In short, FLD stimulates flowering in Arabidopsis by deacetylating the chromatin that surrounds FLC, thereby removing its inhibitory effect on flowering.
Changes in chromatin structure that affect gene expression, such as the mechanisms just described (chromatin remodeling, histone modification, and DNA methylation), are examples of the phenomenon of epigenetics. Epigenetics is defined as alterations to DNA and chromatin structure that affect traits and are passed on to other cells or generations but are not caused by changes in the DNA base sequences. Epigenetic changes and their affect on gene regulation will be discussed in more detail in Chapter 21. ![]() TRY PROBLEM 18
TRY PROBLEM 18
Chromatin Immunoprecipitation
Our understanding of how changes in chromatin structure are associated with gene expression, as well as how DNA binding proteins affect transcription, have been greatly advanced by the use of a technique called chromatin immunoprecipitation (ChIP). This technique allows researchers to determine the specific locations within the genome where proteins interact with DNA. Those proteins might be histones that have undergone modifications, transcription factors, or other proteins that bind to promoters and enhancers (sequences that affect the transcription of distant genes, see Chapter 13).
The basic idea of ChIP is that a particular protein and the DNA to which it is bound are isolated, the protein and DNA are then separated, and the DNA sequence to which the protein was formally bound is identified. The technique has provided a powerful means of determining the genome-wide locations of modified histones and the binding sites for transcription factors and other proteins that affect transcription.
One version of ChIP, called crosslinked ChIP (XChIP) is used for identifying the binding sites of transcription factors and other proteins that bind to DNA. In this procedure (Figure 17.4), the protein and associated DNA are temporarily crosslinked, which means that they are treated with formaldehyde or UV light to create covalent bonds between the DNA and protein. The crosslinking holds the DNA and protein together so that the DNA to which the protein is bound will separate along with the protein. After crosslinking, the cell is lysed and the chromatin is broken into pieces by digestion with an enzyme or by mechanical shearing. Antibodies specific for a particular protein—such as a specific transcription factor—are then applied. The antibodies attach to the protein-DNA complexes and cause them to precipitate. The crosslink is then reversed, separating the DNA and protein. The protein is removed by an enzyme that digests protein but not DNA, leaving fragments of the DNA to which the protein was bound. These fragments can then be sequenced or identified with other methods. The result is the information about the genomic locations of binding sites for the specific protein.
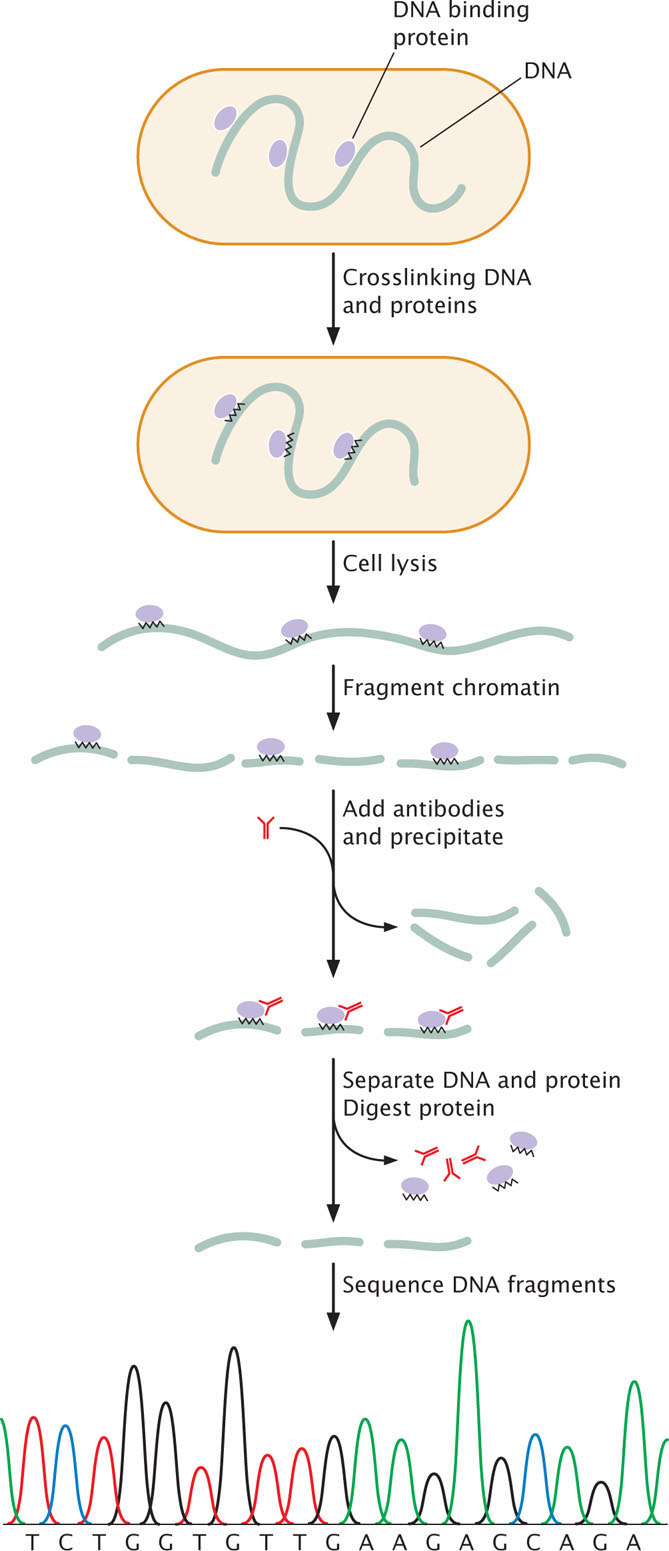
A version of this technique, called native ChIP (nChIP) does not utilize crosslinking. It is often used for finding the locations of modified histone proteins. In this case, crosslinking is not required because the DNA and histone proteins are naturally linked by the nucleosome structure. The chromatin is isolated from the cell and fragmented, and antibodies to a particular protein (usually a specific modified histone) are used to precipitate the protein-DNA complexes. The protein and DNA are separated, the protein digested, and the DNA fragments to which the modified histones were attached are sequenced or otherwise identified.
ChIP analysis has been used to determine the locations of modified histones that activate or repress transcription. As mentioned earlier, the H3K4me3 histone mark is associated with promoters of active genes. nChIP analysis has successfully identified locations of this modified histone in the human genome, helping to identify active promoters in different tissues.
CONCEPTS
The tails of histone proteins are often modified by the addition or removal of phosphate groups, methyl groups, or acetyl groups, or other changes. These modifications alter chromatin structure and affect the transcription of genes.
DNA Methylation
Another change in chromatin structure associated with transcription is the methylation of cytosine bases, which yields 5-methylcytosine (see Figure 10.19). The methylation of cytosine in DNA is distinct from the methylation of histone proteins mentioned earlier. Heavily methylated DNA is associated with the repression of transcription in vertebrates and plants, whereas transcriptionally active DNA is usually unmethylated in these organisms. Abnormal patterns of methylation are also associated with some types of cancer.
DNA methylation is most common on cytosine bases adjacent to guanine nucleotides (CpG, where p represents the phosphate group in the DNA backbone); so two methylated cytosines sit diagonally across from each other on opposing strands:
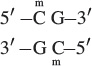
DNA regions with many CpG sequences are called CpG islands and are commonly found near transcription start sites. While genes are not being transcribed, these CpG islands are often methylated, but the methyl groups are removed before the initiation of transcription. CpG methylation is also associated with long-term gene repression, such as on the inactivated X chromosome of female mammals (see Chapter 4).
Evidence indicates that an association exists between DNA methylation and the deacetylation of histones, both of which repress transcription. Certain proteins that bind tightly to methylated CpG sequences form complexes with other proteins that act as histone deacetylases. In other words, methylation appears to attract deacetylases, which remove acetyl groups from the histone tails, stabilizing the nucleosome structure and repressing transcription. The demethylation of DNA allows acetyltransferases to add acetyl groups, disrupting nucleosome structure and permitting transcription. ![]() TRY PROBLEM 19
TRY PROBLEM 19
CONCEPTS
Chromatin structure can be altered by methylation of DNA. In eukaryotes, DNA methylation consists of 5-methylctyosine occurring at CpG dinucleotides. DNA methylation is usually associated with repression of transcription.
 CONCEPT CHECK 1
CONCEPT CHECK 1What are some of different processes that affect gene regulation by altering chromatin structure?