19.4 DNA Sequences Can Be Determined and Analyzed
In addition to cloning and amplifying DNA, molecular techniques are used to analyze DNA molecules through the study and determination of their sequences.
Restriction Fragment Length Polymorphisms
A significant contribution of molecular genetics has been to provide numerous genetic markers that can be used in gene mapping. We learned how these markers are essential to the success of positional cloning. One group of such markers comprises restriction fragment length polymorphisms (RFLPs), which are variations (polymorphisms) in the patterns of fragments produced when DNA molecules are cut with the same restriction enzyme (Figure 19.22). These differences are inherited and can be used in mapping, similar to the way in which allelic differences are used to map conventional genes.
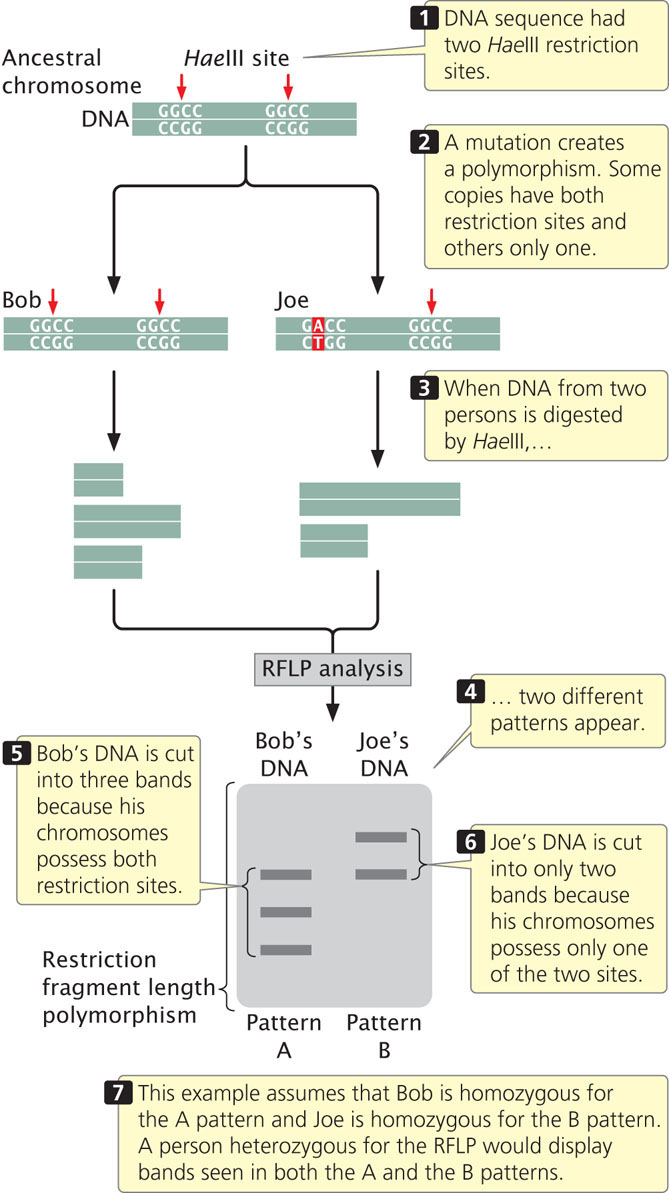
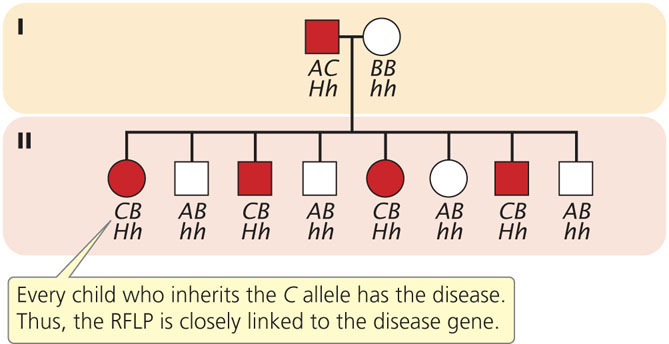
To illustrate mapping with RFLPs, consider Huntington disease, an autosomal dominant disorder. In the family shown in Figure 19.23, the father is heterozygous both for Huntington disease (Hh) and for a restriction pattern (AC). From the father, each child inherits either a Huntington-disease allele (H) or a normal allele (h); any child inheriting the H allele develops the disease, because it is an autosomal dominant disorder. The child also inherits one of the two RFLP alleles from the father, either A or C, which produces the corresponding RFLP pattern. In Figure 19.23, every child who inherits the C pattern from the father also inherits Huntington disease (and therefore the H allele), because the locus for the RFLP is closely linked to the locus for the disease-causing gene. If we had observed no correspondence between the inheritance of the RFLP pattern and the inheritance of the disease, the lack of correspondence would indicate that the genes encoding the RFLP and Huntington disease are assorting independently and are not linked. In recent years, the availability of inexpensive DNA sequencing technology (see below) has decreased the use of RFLPs in gene mapping and genetic diagnosis. Today, the diagnosis of Huntington disease is routinely carried out by amplifying a portion of the Huntington disease gene with PCR. ![]() TRY PROBLEM 41
TRY PROBLEM 41
CONCEPTS
Restriction fragment length polymorphisms are variations in the pattern of fragments produced by restriction enzymes, which reveal variations in DNA sequences. They are used extensively in gene mapping.
DNA Sequencing
A powerful molecular method for analyzing DNA is a technique known as DNA sequencing, which quickly determines the sequence of bases in DNA. Sequencing allows the genetic information in DNA to be read, providing an enormous amount of information about gene structure and function. In the mid-1970s, Frederick Sanger and his colleagues created the dideoxy-sequencing method based on the elongation of DNA by DNA polymerase; at about the same time, Allan Maxam and Walter Gilbert developed a second method based on chemical degradation of DNA. The Sanger method quickly became the standard procedure for sequencing any purified fragment of DNA.
The Sanger, or dideoxy, method of DNA sequencing is based on replication. The fragment to be sequenced is used as a template to make a series of new DNA molecules. In the process, replication is sometimes (but not always) terminated when a specific base is encountered, producing DNA strands of different lengths, each of which ends in the same base.
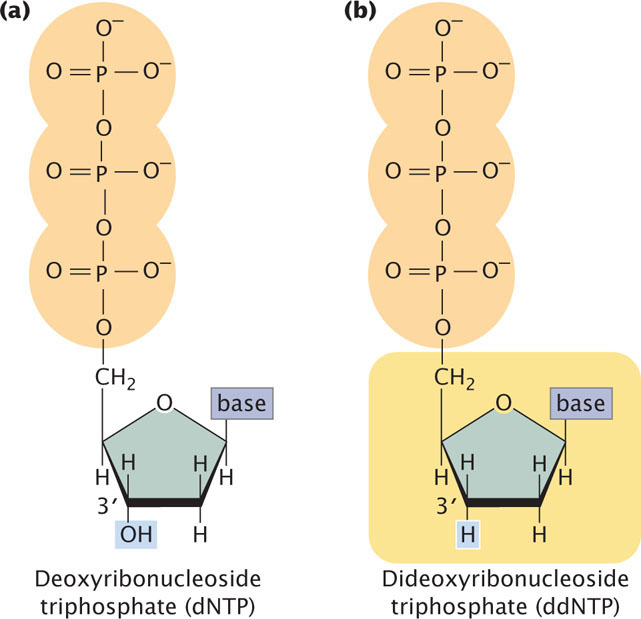
The method relies on the use of a special substrate for DNA synthesis. Normally, DNA is synthesized from deoxyribonucleoside triphosphates (dNTPs), which have an OH group on the 3′-carbon atom (Figure 19.24a). In the Sanger method, a special nucleotide, called a dideoxyribonucleoside triphosphate (ddNTP; Figure 19.24b), is used as one of the substrates. The ddNTPs are identical to dNTPs, except that they lack a 3′-OH group. In the course of DNA synthesis, ddNTPs are incorporated into a growing DNA strand. However, after a ddNTP has been incorporated into the DNA strand, no more nucleotides can be added, because there is no 3′-OH group to form a phosphodiester bond with an incoming nucleotide. Thus, ddNTPs terminate DNA synthesis.
Although the sequencing of a single DNA molecule is technically possible, most sequencing procedures in use today require a considerable amount of DNA; any DNA fragment to be sequenced must first be amplified by PCR or by cloning in bacteria. Copies of the target DNA are isolated and split into four parts (Figure 19.25). Each part is placed in a different tube, to which are added:
- 1. many copies of a primer that is complementary to one end of the target DNA strand;
- 2. all four types of deoxyribonucleoside triphosphates, the normal precursors of DNA synthesis;
- 3. a small amount of one of the four types of dideoxyribonucleoside triphosphates, which will terminate DNA synthesis as soon as it is incorporated into any growing chain (each of the four tubes received a different ddNTP); and
- 4. DNA polymerase.
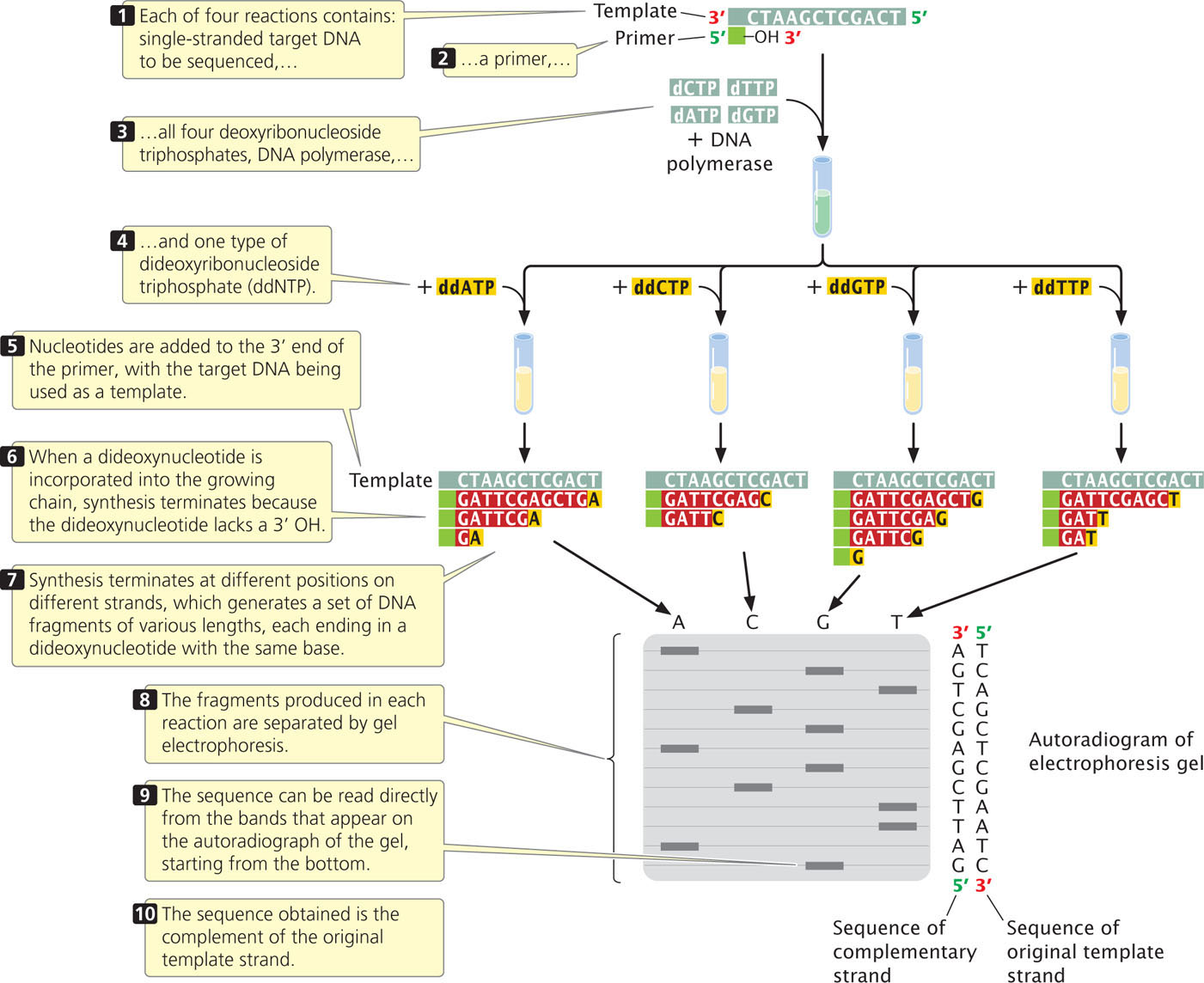
Either the primer or one of the dNTPs is radioactively or chemically labeled so that newly produced DNA can be detected.
Within each of the four tubes, the DNA polymerase enzyme synthesizes DNA. Let’s consider the reaction in one of the four tubes; the one that received ddATP. Within this tube, each of the single strands of target DNA serves as a template for DNA synthesis. The primer pairs to its complementary sequence at one end of each template strand, providing a 3′-OH group for the initiation of DNA synthesis. DNA polymerase elongates a new strand of DNA from this primer. Wherever DNA polymerase encounters a T on the template strand, it uses at random either a dATP or a ddATP to introduce an A in the newly synthesized strand. Because there is more dATP than ddATP in the reaction mixture, dATP is incorporated most often, allowing DNA synthesis to continue. Occasionally, however, ddATP is incorporated into the strand and synthesis terminates. The incorporation of ddA into the new strand occurs randomly at different positions in different copies, producing a set of DNA chains of different lengths (12, 7, and 2 nucleotides long in the example illustrated in Figure 19.25), each ending in a nucleotide that contains adenine.
Equivalent reactions take place in the other three tubes, except that synthesis is terminated at nucleotides with a different base in each tube. After the completion of the polymerization reactions, all of the DNA in the tubes is denatured, and the single-strand products of each reaction are separated by gel electrophoresis.
The contents of the four tubes are separated side by side on an acrylamide gel so that DNA strands differing in length by only a single nucleotide can be distinguished. After electrophoresis, the locations, and therefore the sizes, of the DNA strands in the gel are revealed by the use of radioactive or chemical tags.
Reading the DNA sequence is the simplest and shortest part of the procedure. In Figure 19.25, you can see that the band closest to the bottom of the gel is from the tube that contained the ddGTP reaction, which means that the first nucleotide synthesized had guanine (G). The next band up is from the tube that contained ddATP; so the next nucleotide in the sequence is adenine (A), and so forth. In this way, the sequence is read from the bottom to the top of the gel, with the nucleotides near the bottom corresponding to the 5′ end of the newly synthesized DNA strand and those near the top corresponding to the 3′ end. Keep in mind that the sequence obtained is not that of the target DNA but that of its complement. To see dideoxy sequencing in action, view  Animation 19.3.
Animation 19.3. ![]() TRY PROBLEM 39
TRY PROBLEM 39
For many years, DNA sequencing was done largely by hand and was laborious and expensive. Today, sequencing is usually carried out by automated machines that use fluorescent dyes and laser scanners to sequence thousands of base pairs in a few hours (Figure 19.26). The dideoxy reaction is also used here, but the ddNTPs used in the reaction are labeled with fluorescent dyes, and a different colored dye is used for each type of dideoxynucleotide. In this case, the four sequencing reactions can take place in the same test tube and can be placed in the same well during electrophoresis. The most recently developed sequencing machines carry out electrophoresis in gel-containing capillary tubes. The different-size fragments produced by the sequencing reaction separate within a tube and migrate past a laser beam and detector. As the fragments pass the laser, their fluorescent dyes are activated and the resulting fluorescence is detected by an optical scanner. Each colored dye emits fluorescence of a characteristic wavelength, which is read by the optical scanner. The information is fed into a computer for interpretation, and the results are printed out as a set of peaks on a graph (see Figure 19.26). Automated sequencing machines may contain 96 or more capillary tubes, allowing from 50,000 to 60,000 bp of sequence to be read in a few hours.
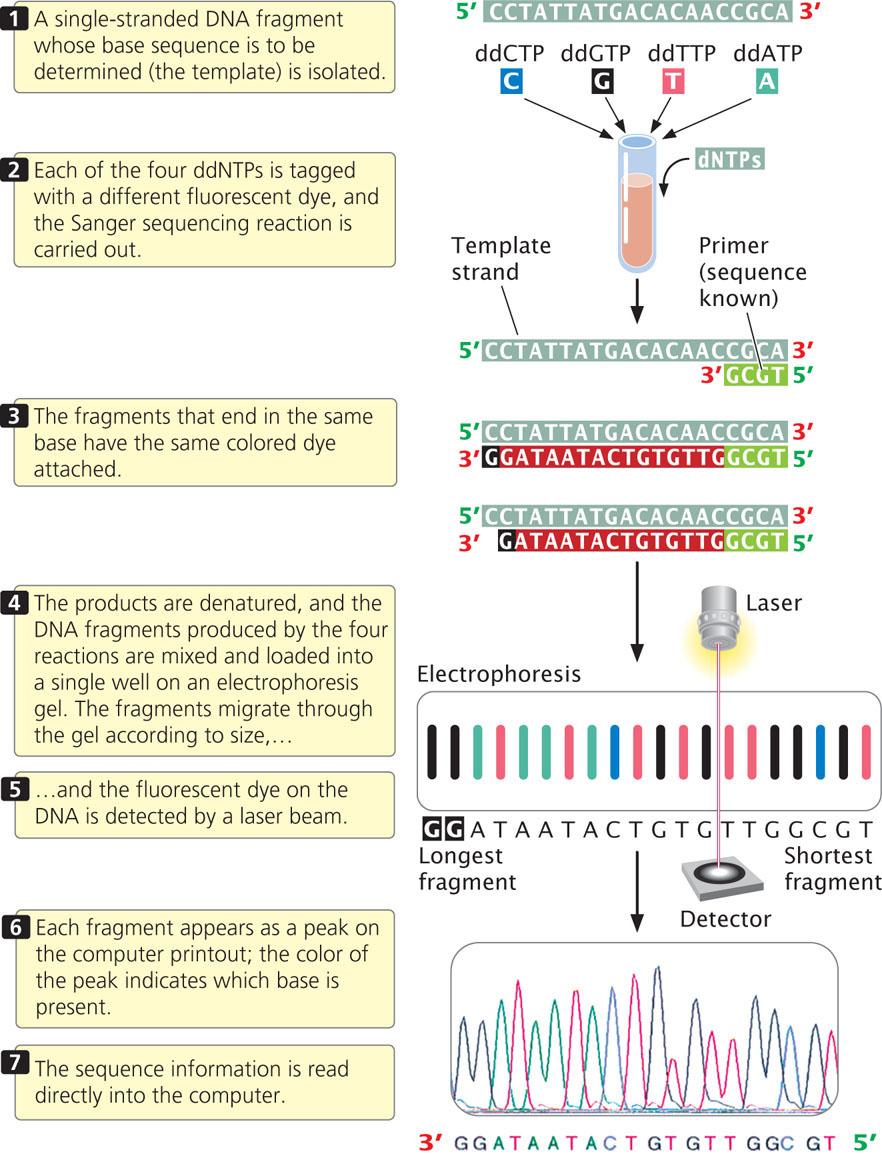
CONCEPTS
The dideoxy sequencing method uses ddNTPs, which terminate DNA synthesis at specific bases.
 CONCEPT CHECK 8
CONCEPT CHECK 8In the dideoxy sequencing reaction, what terminates DNA synthesis at a particular base?
- The absence of a base on the ddNTP halts the DNA polymerase.
- The ddNTP causes a break in the sugar–phosphate backbone.
- DNA polymerase will not incorporate a ddNTP into the growing DNA strand.
- The absence of a 3′-OH group on the ddNTP prevents the addition of another nucleotide.
Next-Generation Sequencing Technologies
Newer methods, called next-generation sequencing technologies, have made sequencing hundreds of times faster and less expensive than the traditional Sanger sequencing method. Most next-generation sequencing technologies do sequencing in parallel, which means that hundreds of thousands or even millions of DNA fragments are simultaneously sequenced.
Pyrosequencing
One type of next generation sequencing, called pyrosequencing, is based on DNA synthesis: nucleotides are added one at a time in the order specified by template DNA and the addition of a particular nucleotide is detected with a flash of light, which is generated as the nucleotide is added.
To carry out pyrosequencing, DNA to be sequenced is first fragmented (Figure 19.27a). Adaptors, consisting of a short string of nucleotides, are added to each fragment. The adaptor provides a known sequence to prime a PCR reaction. The DNA fragments are then made single stranded. In one version of pyrosequencing, each fragment is then attached to a separate bead and surrounded by a droplet of solution (Figure 19.27b). The bead is used to hold the DNA and later deposit it on a plate for the sequencing reaction (see below). Within the droplet, the fragment is then amplified by PCR and the copies of DNA remain attached to the bead. After amplification by PCR, the beads are mixed with DNA polymerase and are deposited on a plate containing more than a million wells (small holes). Each bead is deposited into a different well.
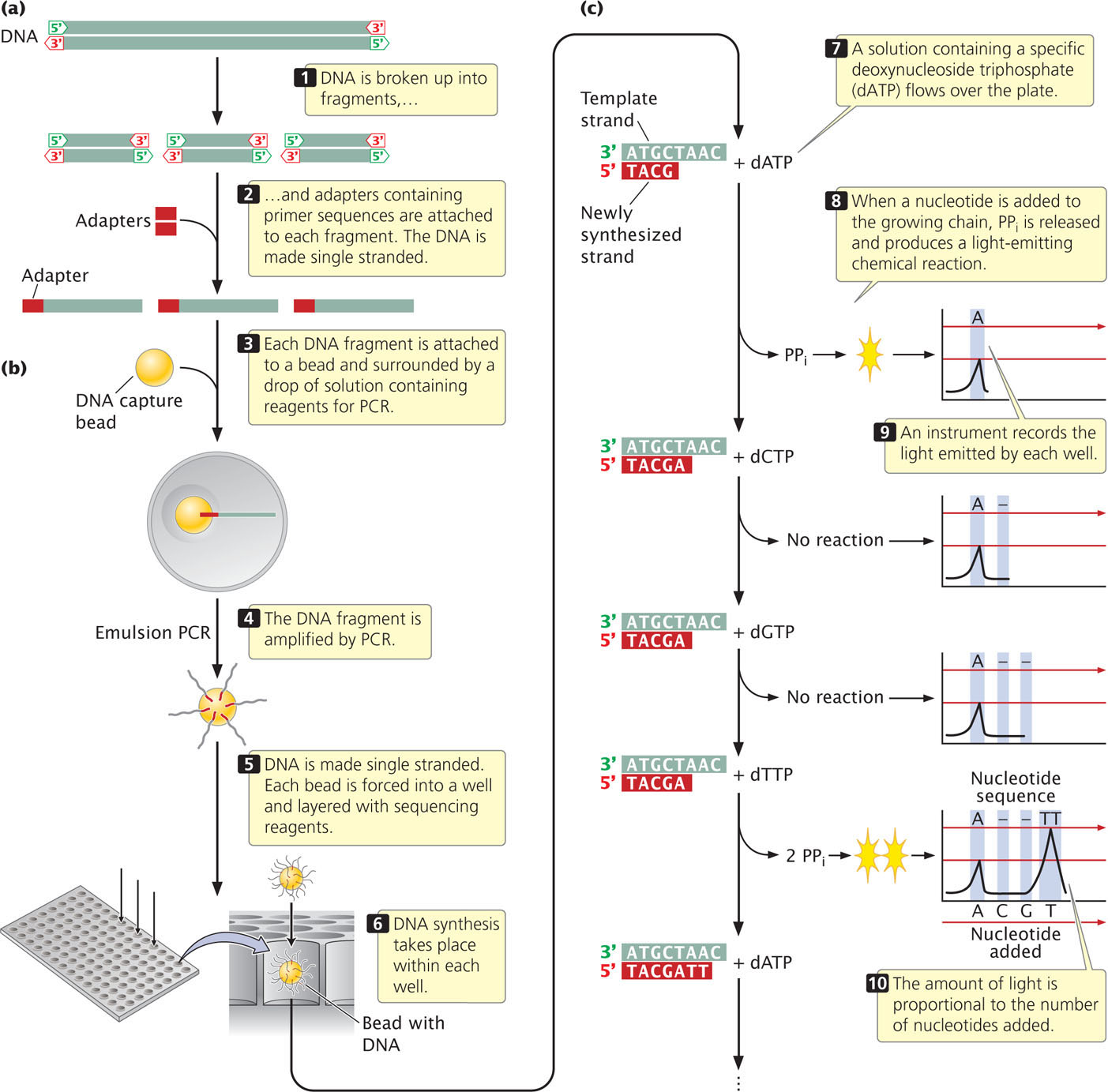
The sequencing reaction takes place in each well and is based on DNA synthesis. Recall from Chapter 12 that the substrate for DNA synthesis is a deoxynucleoside triphosphate, consisting of a deoxyribose sugar attached to a base and three phosphates. In the process of DNA synthesis, two phosphates (pyrophosphate) are cleaved off, and the resulting nucleotide is attached to the 3 end of the growing DNA chain. A solution containing one particular type of deoxynucleoside triphosphate—say, deoxyadenosine triphosphate—is passed across the wells (Figure 19.27c). If the template within a particular well specifies an adenine nucleotide in the next position of the growing chain, then pyrophosphate is cleaved from the nucleoside triphosphate and the adenine nucleotide is added. A chemical reaction uses the pyrophosphate produced in the reaction to generate a flash of light, which is measured by an optical detector. The amount of light emitted in each well is proportional to the number of nucleotides added: if the template in a well specifies three successive adenine nucleotides (As), then three nucleotides are added and three times more light is emitted than if a single A were added. If the position in the template specifies a base other than adenine, no nucleotide is added, no pyrophosphate is produced, and no light is emitted.
As mentioned, the first solution passed over the plate contains adenosine triphosphate and allows adenine nucleotides to be added to the template. Each well with a template that specifies adenine in the next position will generate a flash of light. Then, a solution with a different type of nucleoside triphosphate—say, deoxyguanosine triphosphate—is passed across the wells. Any fragment that specifies a G in the next position of its growing chain will add a guanine nucleotide and emit a flash of light. The nucleotide triphosphates are passed across the wells in a predetermined order, and the light emitted by each well is measured. In this way, hundreds of thousands or millions of fragments of DNA are sequenced simultaneously on the basis of the order in which nucleotides are added to the 3′ end of the growing chain. Pyrosequencing determines only the sequence of the fragments in each well; it does not, by itself, allow the sequences of these fragments to be reassembled into the sequence of the entire original piece of DNA. The reassembly of sequenced fragments into a continuous stretch of DNA is a general problem in the sequencing of genomes and will be explained in Chapter 20.
Illumina Sequencing
Several other forms of next-generation sequencing are widely used. Illumina sequencing employs a technology similar to that of the Sanger dideoxy method. Special nucleotides are used that have a fluorescent tag attached, with a different colored tag for each type of nucleotide. Each nucleotide also has a chemical group (a terminator) that, once incorporated into the growing DNA chain, prevents the incorporation of any additional nucleotides. This is similar to termination caused by dideoxynucleotides in Sanger sequencing. However, here the terminator is reversible—it can be chemically removed. To carry out sequencing, the DNA is first fragmented into millions of short overlapping fragments. The fragments are attached to a slide and then amplified, creating clusters of up to 1000 copies of each fragment in close proximity on the slide. The fragments are then denatured and a solution of primers, DNA polymerase, and the special nucleotides are added. The primer attaches to each DNA template and the first nucleotide is incorporated into the newly synthesized strand. The solution is washed away, and the tag on the incorporated nucleotide is excited with a laser, which causes it to fluoresce. As mentioned above, each type of nucleotide (A, T, G, or C) has a different colored fluorescent tag, so the color of the light produced reveals the type of the nucleotide just added. The terminator and the fluorescent tag are then chemically removed and the process is repeated. As the nucleotides are added one at a time, the sequence is read as a series of flashes of colored light from each cluster of DNA. Hundreds of thousands of DNA clusters, each consisting of copies of a different DNA fragment, are sequenced simultaneously, allowing large amounts of DNA to be sequenced in a short time.
Most next-generation sequencing techniques read shorter DNA fragments than the Sanger sequencing reactions do, but because hundreds of thousands or millions of fragments are sequenced simultaneously, these methods are much faster than traditional Sanger sequencing technology.
Third-Generation Sequencing Technology
Even more advanced and rapid sequencing methods, typically called third-generation sequencing, are currently under development. For example, nanopore sequencing determines the sequence of individual molecules of DNA. In this method, a single strand of DNA is passed through a tiny hole—a nanopore—in a membrane. As the molecule passes through the nanopore, it disrupts an electrical current in the membrane, and the nature of the disruption is affected by the shape of the molecule passing through the nanopore. Each of the four bases of DNA causes a characteristic disruption, so the sequence of DNA can be read by analyzing the membrane current as the strand passes, one nucleotide at a time, through the nanopore. Hundreds of thousands of nanopores can be created on a single chip, so that many DNA fragments can be read simultaneously. One of the goals of third-generation sequencing technology is to develop a method that can sequence an entire human genome for less than $1000.
CONCEPTS
New next- and third-generation sequencing methods sequence many DNA fragments simultaneously and provide a much faster and less-expensive determination of a DNA base sequence than does the Sanger sequencing method.
DNA Fingerprinting
The use of DNA sequences to identify individual persons is called DNA fingerprinting or DNA profiling. Because some parts of the genome are highly variable, each person’s DNA sequence is unique and, like a traditional fingerprint, provides a distinctive characteristic that allows identification.
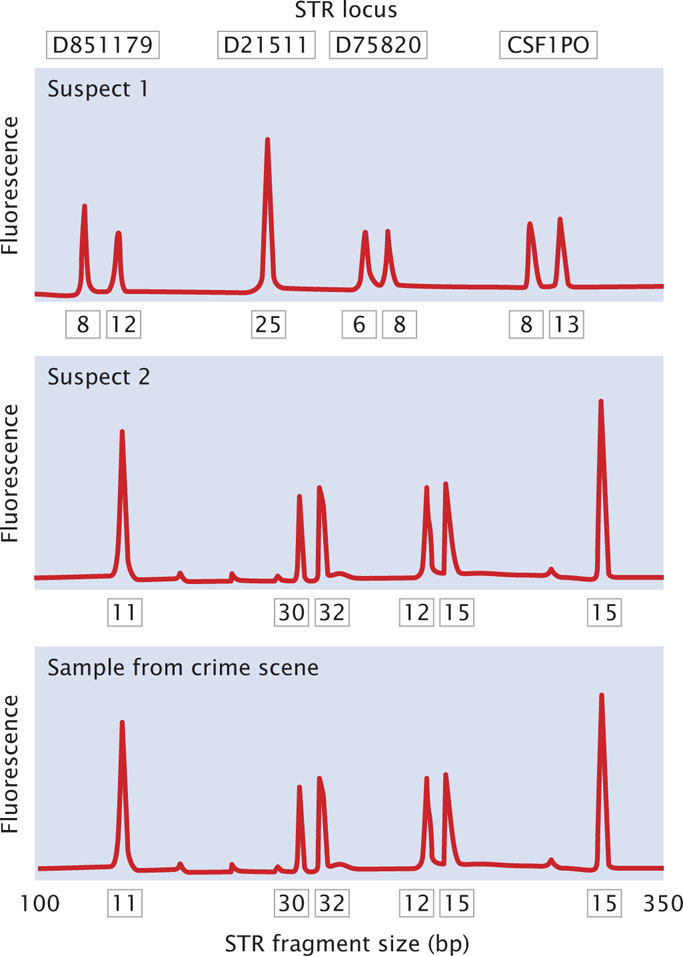
Today, most DNA fingerprinting utilizes microsatellites, or short tandem repeats (STRs), which are very short DNA sequences repeated in tandem (see Chapter 11). These repeated sequences are found at many loci throughout the human genome. People vary in the number of copies of repeat sequences that they possess at each locus. The STRs are typically detected with PCR, using primers flanking the microsatellite repeats so that a DNA fragment containing the repeated sequences is amplified (Figure 19.28). The length of the amplified segment depends on the number of repeats; DNA from a person with more repeats will produce a longer amplified segment than will DNA from a person with fewer repeats.
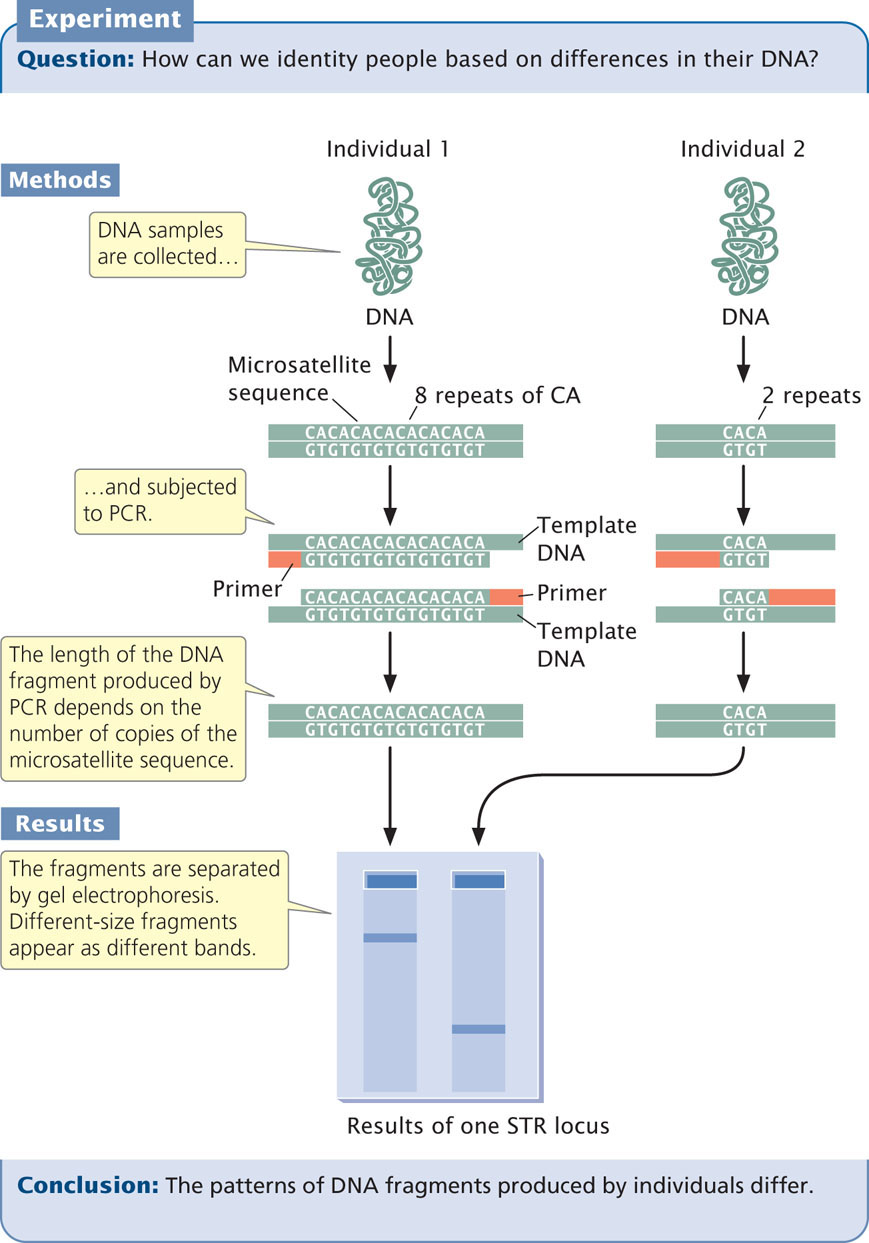
The primers used in the PCR reaction are tagged with a fluorescent label so that the resulting DNA fragments can be detected with a laser. Primers for different STR loci are labeled with different colored primers, so that similar-sized products of different loci can be differentiated. After PCR, the fragments are separated on a gel or by a capillary electrophoresis machine. In capillary electrophoresis, the presence of each fragment is detected as it migrates past a laser, and a computer then calculates the size of each fragment based on its rate of migration. The fragments are represented as peaks on a graph; the distance on the horizontal axis represents the size of the fragment, while the height of the peak represents the amount of DNA (Figure 19.29). Homozygotes for an STR allele have a single tall peak; heterozygotes have two shorter peaks. When several different microsatellite loci are examined, the probability that two people have the same set of patterns becomes vanishingly small, unless they are identical twins.
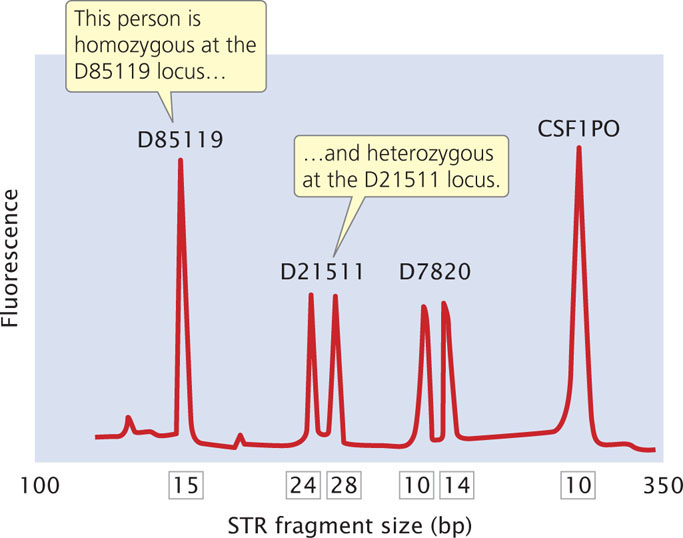
The Federal Bureau of Investigation has developed a system using 13 STR loci (Table 19.3) that are commonly utilized for identifying people and in solving crimes. These loci make up the Combined DNA Index System (CODIS). Each STR locus in CODIS has a large number of alleles and is located on a different human chromosome, and so variation at each locus assorts independently. When all 13 CODIS loci are used together, the probability of two randomly selected people having the same DNA profile is less than 1 in 10 billion.
| Locus Name | Chromosome | Number of Repeats | Number of Alleles* |
|---|---|---|---|
| CSF1PO | 5 | 5-17 | 10 |
| FGA | 4 | 12-51 | 23 |
| TH01 | 11 | 3-14 | 8 |
| TPOX | 2 | 4-16 | 8 |
| VWA | 12 | 10-25 | 10 |
| D3S1358 | 3 | 6-26 | 10 |
| D5S818 | 5 | 4-29 | 9 |
| D7S820 | 7 | 5-16 | 11 |
| D8S1179 | 8 | 6-20 | 11 |
| D13S317 | 13 | 5-17 | 8 |
| D16S539 | 16 | 4-17 | 7 |
| D18S51 | 18 | 5-40 | 19 |
| D21S11 | 21 | 12-43 | 22 |
|
*US population.
Source: J. M. Butler and C. R. Hill, Forensic Science Review 24:15-26, 2012. |
|||
In a typical application, DNA fingerprinting might be used to confirm that a suspect was present at the scene of a crime. A sample of DNA from blood, semen, hair, or other body tissue is collected from the crime scene. If the sample is very small, PCR can be used to amplify it so that enough DNA is available for testing. Additional DNA samples are collected from one or more suspects. The pattern of DNA fragments produced by DNA fingerprinting from the sample is then compared with the patterns produced by DNA fingerprinting of the DNA from the suspect. A match between the samples can provide evidence that the suspect was present at the scene of the crime (Figure 19.30).
Since its introduction in the 1980s, DNA fingerprinting has helped convict a number of suspects in murder and rape cases. Suspects in other cases have been proved innocent when their DNA failed to match that from the crime scenes. Initially, calculating the odds of a match (the probability that two people could have the same pattern) was controversial, and there were concerns about quality control (such as the accidental contamination of samples and the reproducibility of results) in laboratories where DNA analysis is done. Today, DNA fingerprinting has become an important tool in forensic investigations. In addition to its application in solving crimes, DNA fingerprinting is used to assess paternity, study genetic relationships among individual organisms in natural populations, identify specific strains of pathogenic bacteria, and identify human remains.
CONCEPTS
DNA fingerprinting detects genetic differences among people by analyzing highly variable regions of chromosomes.
CONCEPT CHECK 9How are microsatellites detected?
Application: Identifying People Who Died in the Collapse of the World Trade Center
On the morning of September 11, 2001, terrorists hijacked and flew two passenger planes into the World Trade Center towers in New York City. The catastrophic damage and ensuing fire led, within a few hours, to the complete collapse of all 110 floors of both towers, killing almost 3000 building occupants and rescue personnel. The tremendous destructive force generated by the collapse of the towers, with their 425,000 cubic yards of concrete and 200,000 tons of steel, pulverized many of the bodies beyond recognition.
In the days immediately following the World Trade Center collapse, forensic scientists began the task of identifying the remains of those who perished. The goal was to provide evidence for the ongoing criminal investigation of the attack and to identify the remains for families and friends of the victims. This task was unprecedented in scope and difficulty. There was no complete list of victims (such as a passenger list in an airline crash) with which investigators could match the remains. In all, almost 20,000 individual remains were found, varying from whole bodies to tiny fragments of charred bone. The remains were subjected to fires with temperatures exceeding 1000°C that burned for more than 3 months. The collapse of the buildings intermixed many victims’ remains, and many body fragments were not recovered for months, during which time they were exposed to dust, water, bacteria, and decay.
The usual means of victim identification—personal items, fingerprints, dental records—were of little use for most of the World Trade Center remains. Identification of the majority of the remains was made with the use of DNA fingerprinting (Figure 19.31).

DNA was first extracted from the tissue samples by using sterile techniques to prevent cross-contamination between samples. After the DNA had been extracted, PCR was used to amplify STR loci in the CODIS system (see previous section). The DNA fingerprint generated from each body sample was compared with that of DNA extracted from reference samples, such as the victims’ toothbrushes and blood samples, provided by families and friends. If the DNA from a body part had the same alleles at all 13 loci as in the reference sample, then a positive identification was made. When no reference sample was available, investigators collected DNA from family members and tried to match the DNA profiles of remains to those of relatives; in this case, some, but not all STR alleles would match.
Unfortunately, many of the remains were so badly degraded that little DNA remained and one or more of the STR loci could not be amplified. For these remains, DNA fingerprinting was also carried out on mitochondrial DNA (see Chapter 11). Because there are many mitochondria per cell and each mitochondrion contains multiple DNA molecules, there are many more copies of mitochondrial DNA per cell than nuclear DNA; mitochondrial DNA has been successfully extracted and analyzed from ancient remains, such as Neanderthals (see introduction to Chapter 10). Alone, fingerprinting from mitochondrial DNA was insufficient to provide identification with a high degree of confidence (because there are not as many sequences that vary among people as in the CODIS loci) but, when it was used in conjunction with data from at least some STR loci, a positive identification could often be made.
Used in combination, these techniques allowed the remains of many victims to be positively identified. However, despite the heroic efforts of hundreds of molecular geneticists, forensic anthropologists, and medical examiners, no positively identified remains were recovered for almost half of the people who are thought to have died in the disaster.