21.4 The Epigenome
In 2003 researchers declared that essentially the entire human genome had been sequenced. This monumental achievement provided a wealth of information about how genetic information is encoded within the genome. Yet, the DNA base sequence is only a partial record of the heritable information. As we have discussed, additional epigenetic information is contained within chromatin structure: information that is heritable and affects how the base sequence is expressed. The overall pattern of chromatin modifications possessed by each individual organism has been termed the epigenome. Over the past few years, techniques have become available for detecting and describing epigenetic modifications across the genome.
Detecting DNA Methylation
A number of techniques have been developed for examining levels of DNA methylation. Some of these rely on restriction endonucleases, enzymes that make double-stranded cuts in the DNA at specific base sequences (see Chapter 19). Some restriction enzymes are sensitive to methylation and will not cut a sequence that contains 5-methylcytosine, whereas other restriction enzymes are insensitive to methylation. By cutting DNA with enzymes that are sensitive to methylation and with enzymes that are not, and then analyzing the resulting fragments, overall patterns of methylation can be determined.
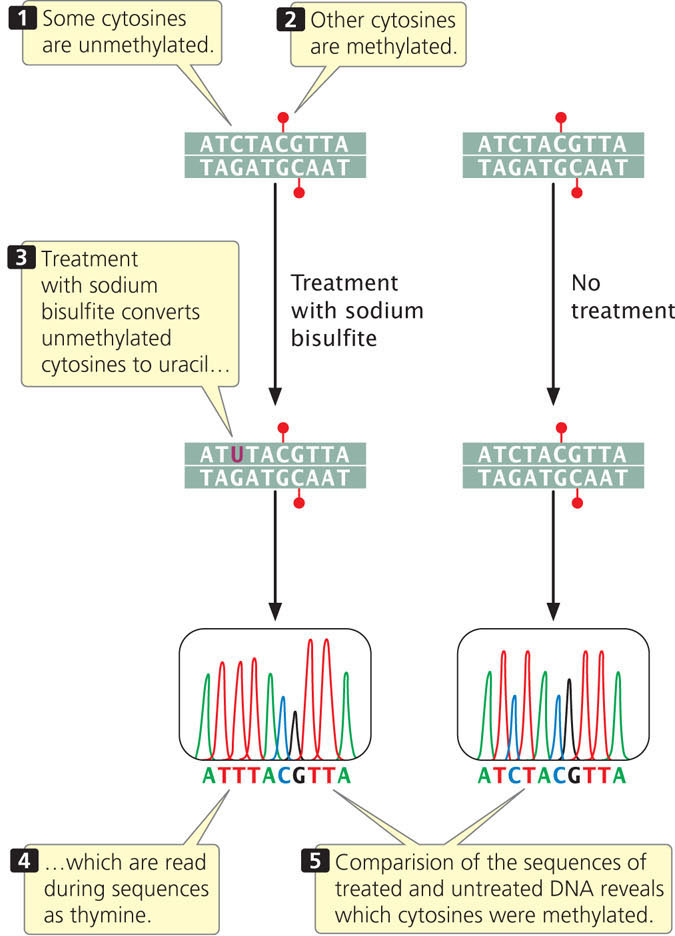
A more precise and widely used technique for analyzing DNA methylation is bisulfite sequencing (Figure 21.13). In this technique, genomic DNA is first treated with sodium bisulfite, which chemically converts unmethylated cytosine to uracil. Uracils are then detected as thymine during sequencing. However, 5-methylcytosine is not chemically altered by treatment with bisulfite and is detected as cytosine during sequencing (see Chapter 19 for a discussion of DNA sequencing). By sequencing genomic DNA with and without bisulfite treatment, researchers are able to determine the locations of all copies of 5-methylcytosine in the DNA.
Detecting Histone Modifications
Histone modifications can be detected by breaking the chromatin into fragments and applying an antibody specific to a particular histone modification, a process called chromatin immunoprecipitation (abbreviated ChIP; see Chapter 17). The antibody causes the chromatin with the histone modification to precipitate and separate from chromatin fragments without the modification. The protein is then removed by digestion with an enzyme that degrades protein but not DNA, and the DNA fragment with which the histone was associated is sequenced. This provides information about where in the genome the histone modifications occur.
Genome-Wide Epigenetic Marks
Using these techniques, geneticists have compared the epigenomes of different types of cells. For example, researchers determined the distribution of 5-methylcytosine across the entire genome in two cell types: (1) an undifferentiated human stem cell; and (2) a fibroblast. The researchers found widespread differences in the methylation patterns of these cells. The finding that patterns of DNA methylation vary among cell types supports the idea that cytosine methylation provides the means by which cell types stably maintain their differences during development. Other researchers have compared the epigenomes of cancer cells and normal cells and observed distinct epigenetic marks associated with cancer.
628
Similarly, researchers have mapped the genomic locations of histone modifications in different cell types. These studies detected specific histone modifications associated with promoters and enhancers of active genes. In one study, researchers mapped nine different epigenetic marks in nine different types of human cells. They were able to determine how the chromatin marks varied across cell types and compared the epigenetic marks associated with active and repressed genes. Because specific epigenetic marks are often associated with regulatory elements such as promoters and enhancers, researchers have used the presence of these marks to map the locations of these regulatory elements throughout the genome.
CONCEPTS
The epigenome is the complete set of chromatin modifications possessed by an individual organism.